Abstract
- 邻位技术利用高度简化的NGS工作流程,实现在流动槽上的文库制备,在测序前完全消除了标准文库制备的过程
- 标准簇生成和 SBS 测序与DRAGEN算法中的簇邻近度信息相结合,以解锁远距离信息
- 早期测试表明其可以增强挑战性基因组区域的映射、实现超长定相并改善大型结构重排的检测
- 首款商业化产品预计将于 2026 年推出,将通过现有的NovaSeq X系统实现具有成本效益的、人类全基因组
简介
新一代测序方法,主要是合成测序 (SBS),在过去 25 年里取得了显著进步,使其成为基因组应用中广泛使用的一种技术。虽然因美纳短读长全基因组测序 (srWGS) 在大多数人类基因组上实现了高度准确的覆盖,1但基因组的特定区域仍然难以解析。此外,一些类别的基因组变异,包括大型结构重排,难以使用标准短读长进行检测。更长的读取长度具有解决其中一些挑战的能力,但相对成本较高且具难以扩展,需要高输入量和严格的样本输入要求。
在这里,我们引入了邻位映射读技术,这是一种利用流动槽上文库制备和信息学的创新方法,利用来自相邻纳米井中的簇的邻近信息,使用标准 SBS 测序生成远程基因组见解。邻位技术保持了标准 SBS 的准确性、覆盖深度和可扩展性,同时增加了定相、增强了可映射性,并改善了通常与长读长方法相关的结构变异检测。这种新颖的方法为全面的全基因组分析提供了强大、可访问的解决方案。
请参阅本文末尾的术语表。
全新技术
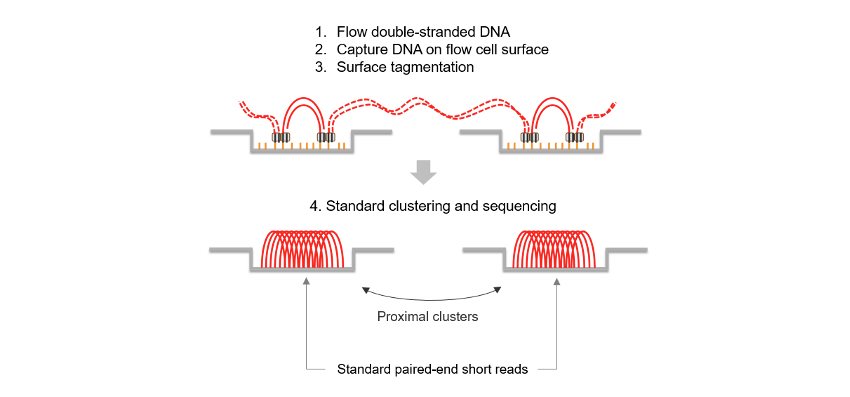
邻位技术与其他通过标准短读长测序(例如Hi-C或链接读取)生成长距离信息的方法不同。邻位技术通过将提取的 DNA 直接应用于流动槽表面来消除手动文库制备,流动槽表面结合的转座酶可以进行原位切割标记。这种流动槽上的文库制备可确保样本基因组中的相邻区域在流动槽上保持物理邻近。邻位技术不依赖于复杂的标记、分子条形码或长连续读取,而是利用相邻簇的空间邻近度,使用标准 SBS 测序从样本基因组中解锁远程基因组信息,从而:
- 在难以映射的区域改进了映射和变异识别
- 超长相位,高达数兆碱基
- 改进了大型 (> 50 bp) 结构重排的识别
工作原理
高度简化的、直接在流动槽上完成的工作流程
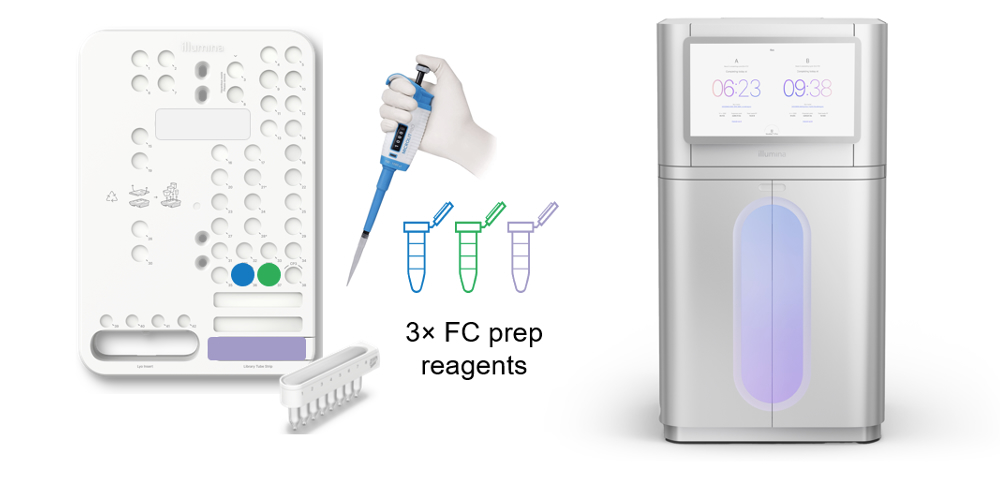
邻位工作流程始于一种新型的流动槽上的文库制备,使用基于转座体的文库制备流程,只需低 DNA 输入和 NovaSeq X系列的高测序质量。实验流程不需要对测序仪进行任何修改,只需自定义测序配方,即可在现有大量已安装仪器上使用。
邻位实验工作流程:
- 将提取的 DNA 模板添加到文库条带管中
- 将专用试剂添加到测序卡盒的定制引物孔中
- 加载耗材并启动运行
邻位读取映射和邻近度分析
邻位技术的优势远远超出了工作流程的简化。通过利用邻近信息,来自相邻簇的读取段被重建为原始DNA模板分子的交错版本。这展示在图3中,其中每个节点代表了一个来自于流动槽簇的读取配对,它们之间的连线提示了基于流动槽和基因组邻近度的组合所预测的它们之间的关联。图3进一步放大了这种交错显示,标明了来自相同原始模板分子的读取段之间的基因组距离,其中模板长度超过300 kb。这些关联是通过临近性模型得出的,这一模型提供了Phred格式的质量分数,描述两个读取段落入某一流动槽位置和给定基因组距离内的概率。分数越高,两个读取段越有可能来自相同的模板分子。这一特性是邻位技术独有的,在任何其他 NGS 分析都不具备这一特性。来自相同模板分子的读取段也具有相同的单倍型。这种 Phred格式的邻近质量和一般邻近特征的组合被用于DRAGEN 二级分析,将读取段分配给难以映射的区域、提取定相信息和识别变异。
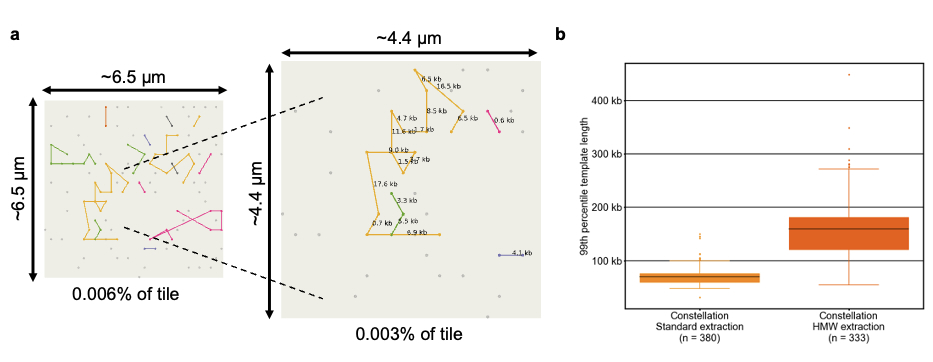
图 3a:从流动槽生成的簇被组织成不同的模板,以不同的颜色呈现。在每个模板内,基于其基因组坐标,这些簇被依序排列。配对读取段之间的连接用其对应的基因组距离进行注释。
图3b:箱形图显示了标准和 高分子量(HMW) 提取中所获得的模板大小(99百分位范围)
邻位技术的优势
提高难以映射区域的性能
在基因组的有限区域中,由于高度同源性或其它重复背景,实现标准短读取段的唯一映射具有挑战性,这使得难以区分多个候选映射位置。邻位读映射使用来自相邻簇的邻近信息的确可以进行唯一映射,以将读取段分配到正确的基因组位置。
应用邻近度信息可以更自信地绘制基因组图谱并全面覆盖基因组,包括难以映射的临床相关基因,如 STRC 和 PMS2(图 5 和图 6)
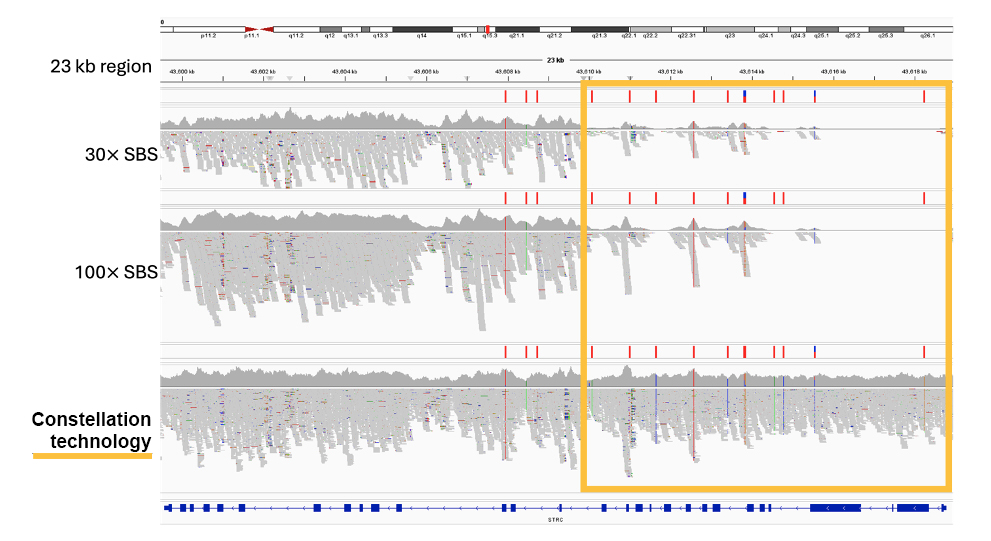
图 5:使用邻位技术恢复 STRC 基因的覆盖度。STRC 具有假基因 STRCP1,序列相似性 > 99%,使得使用标准全基因组文库制备进行映射具有挑战性。STRC 中的一些突变与儿童的非综合征性听力损失有关。
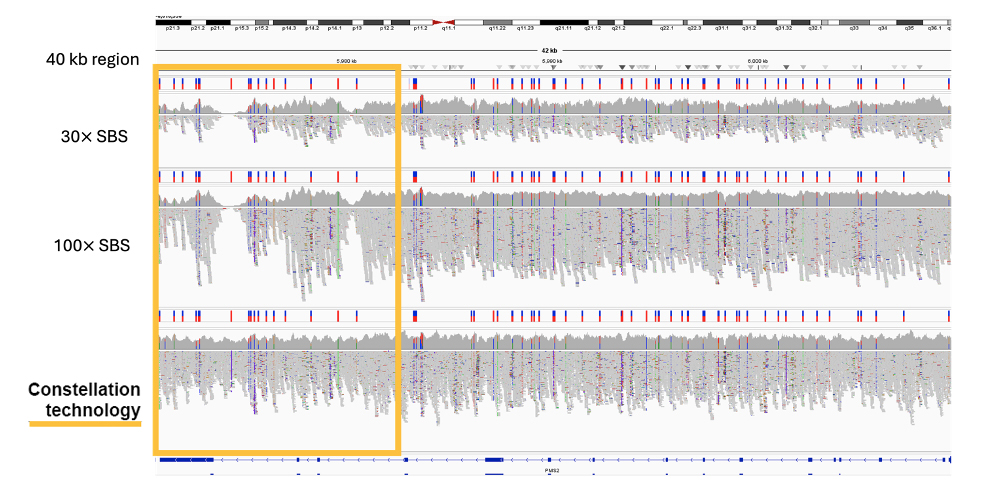
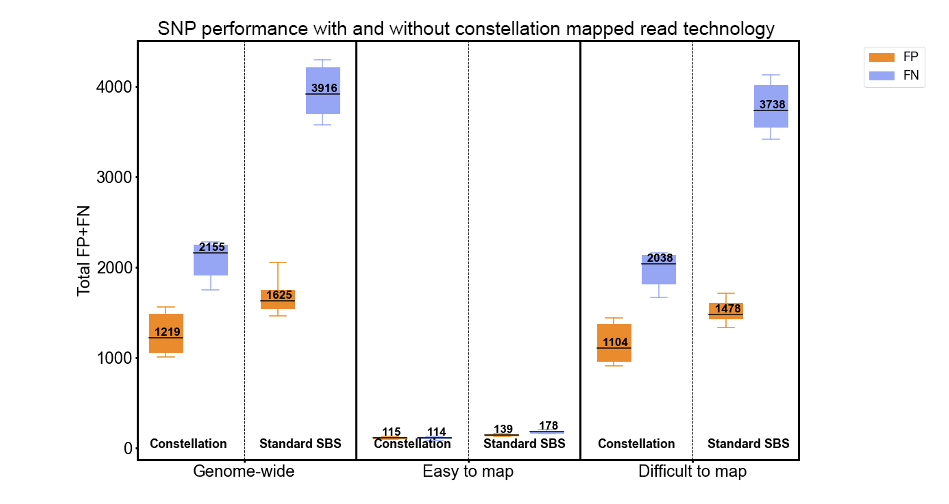
图 7:使用 rtgeval 对11次HG002邻位读取运行的SNP 变异识别性能进行评估,对比NIST瓶中基因组(GIAB) v4.2.1 真值集(truth set)。难以映射和易于映射的区分由 瓶中基因组(GIAB) v 3.3的GRCh38_alldifficultregions.bed.gz 和GRCh38_notinalldifficultregions.bed.gz所定义。
超长定相
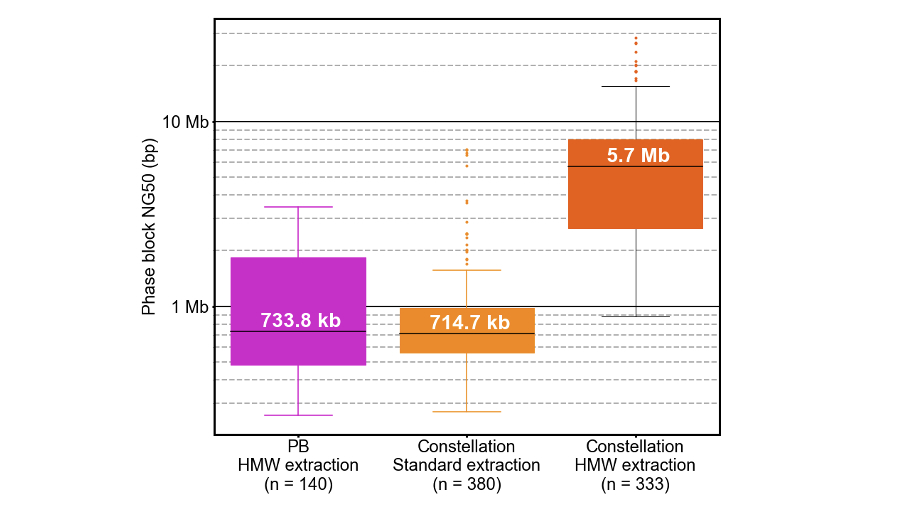
定相测序通过定义单倍型、识别复合杂合子,能够获得更深入的见解。使用邻位技术的定相特别强大,因为它的能力仅由流动槽上捕获的天然DNA模板长度所界定,而不是读取长度,目前可从数百Kb延伸到数Mb。保留较大模板的高分子量 (HMW) 提取方法被证明有助于实现更大的定相块。
使用标准 DNA 提取时,邻位定相块 NG50约为 715 kb,使用 HMW DNA 提取时约为 5.7 Mb(图 8)。初步测试表明,通过标准 DNA 提取,邻位技术可完全定相约85%的基因(中位数),而通过 HMW DNA 提取则可以实现约95%基因的完全定相(中位数)。此外,在标准和 HMW DNA 提取中,邻位技术可对所有杂合 SN的约98%进行定相。
提升结构变异识别
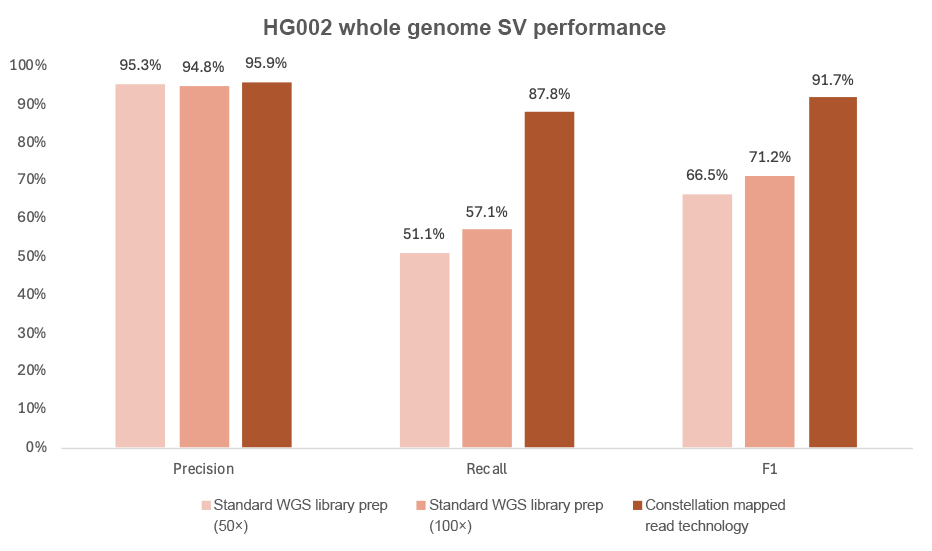
邻位技术具有改善的结构变异(> 50 bp)识别的额外获益。使用 DRAGEN v4.3 二级分析,邻位技术显示了 结构变异(SV) 识别的显著改善,从使用标准 SBS 的 51.5% 提高到 87.8%(图 9)。
随着邻位技术和定制变异识别方法的进一步开发,我们预计在读取映射和小变异及大变异识别方面将有进一步的提升。
基因组结构的新型可视化
邻位映射读取技术解决大型结构重排的能力超越了传统变异识别性能的基准。通过从基因组的任何一对区域之间的邻近簇提取关于读取段的信息,基因组结构图的高分辨率视觉表达成为可能,这被称为“共位图”。
这些图是通过将基因组分成区块(bin),并确定每个可能的基因组区块配对的相邻簇中的读取段数量来生成的。当某对区块中来自邻近簇的读取数量较高时,这通常表明这些区块在基因组中具有较近的邻近关系。在没有结构变异的区域中,参考基因组中相邻的基因组区块在样本中也相邻,因此在共位图中显示为对角线。当一个区域内存在结构变异时,参考基因组中附近的基因组区块在样品中不再靠近,因此表现出各种非对角线信号。
图10提供了与X染色体上的F8基因重叠区域的这些共位图的实例,其中图10a示意了不存在SV的样本,而图10b展示了内含子22倒置(inversion)的样本。
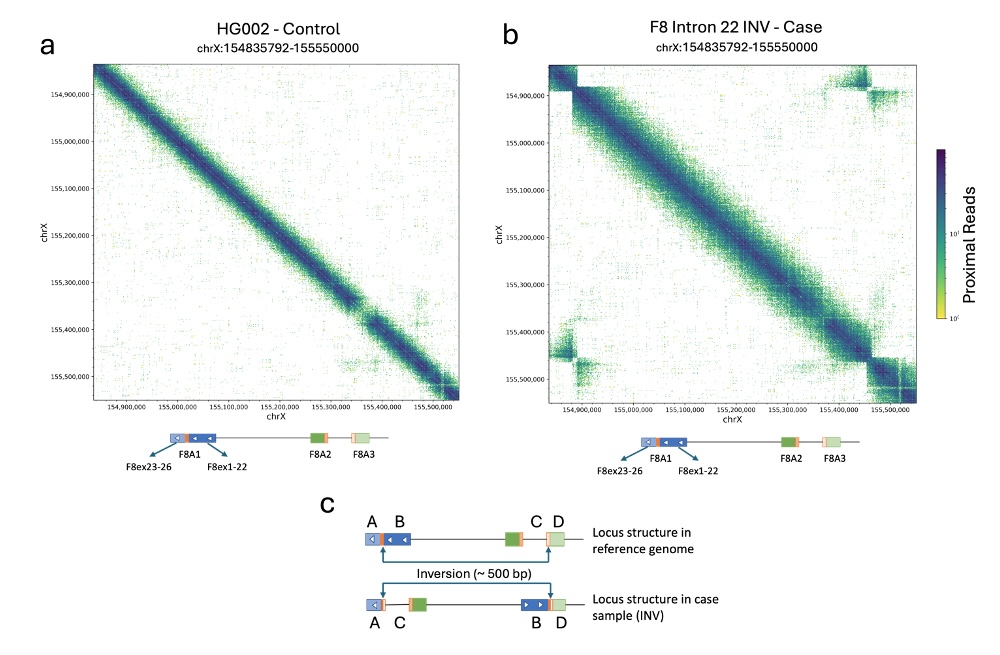
图10a:HG002细胞系X染色体上F8基因位点的共位图
图10b:内含子22倒置的DNA样本的X染色体上的F8基因座的共位图。在F8A3上游的F8外显子23-26之间以及F8A3下游的F8外显子1-22之间的非对角线信号的累积F8A3提示了在内含子22和F8A3之间存在边界的倒置。
图10c:图示参考基因组中以及具有致病性倒置的样品中的F8基因座结构。当存在倒置时,在区域A(F8外显子23-26)与C(F8A3的上游)之间以及区域B(F8外显子1-22)与D(F8A3的下游)之间观察到新的信号。
也可以在携带倒置的样本中观察到对角线中缺乏信号,这表明倒置边界的任一侧的区域在案例样本中是彼此远离的。
图10b中显示的倒置事件在F8的22号内含子的片段重复中具有一个边界,并且在F8A3中的对应片段重复中具有另一个边界(约500 kb远)。片段重复的长度约为 10 kb,呈反向,并且具有 > 99.7% 的序列相似性。这些特性使得通过标准短读长测序无法检测到这一倒置。
共位图能够检测和可视化复杂的平衡和非平衡结构重排,即使事件边界发生在基因组难以映射的区域中。
结论和后续
这只是开始。
邻位映射读技术是一种功能强大的创新底层技术,具有广泛的功能。我们在这里展示了它在人类基因组测序方面的一些优势,但未来的多个应用还在评估中。基于邻位技术的首款商业化产品定于 2026 年上半年推出,并将利用现有的 NovaSeq X 系列测序仪为全面的人类 WGS 创造一个可访问、经济高效的解决方案。
请点击此链接查看 Illumina 首席技术官 Steve Barnard 和 Broad Clinical Labs 首席科学官 Niall Lennon 的 ASHG 2024的演示和早期成果展示。
在下方注册,了解邻位映射读取技术和未来产品的发展动态。
了解更多信息
参考资料
- Behera S、Catreux S、Rossi M等人使用DRAGEN进行全面的基因组分析和大规模变异检测。Nat Biotechnol。2024 年 10 月 25 日在线发布:1-15. doi:10.1038/s41587-024-02382-1
术语表
| 术语 | 定义 |
|---|---|
模板分子 |
从样品中提取的大分子连续双链DNA分子 |
标准 WGS |
使用手动文库制备和标准 SBS 测序进行全基因组测序 |
酶切打断Tagmentation |
使用转座酶切割 DNA 片段和添加接头序列(标记)的过程 |
转座酶Transposome |
以二聚体形式存在的DNA转座酶复合体 |
簇 |
流动槽上的DNA扩增点,之后将被测序 |
邻近簇proximal cluster |
在流动槽上彼此物理靠近的簇。 |
定相块NG50 phase block N50 |
一旦50%的目标区域(基因组或其它)已经被定相,这时的定相块的长度。请注意,无法对给定目标区域的 50% 进行定相分析的技术的 NG50为0bp。 |
可完全定相的基因百分比 |
来自特定来源(例如,NCBI RefSeq、ENCODE、MANE)的基因区域完全包含在单个定相块中的百分比。 |
可定相的杂合变异百分比 |
定相杂合小变异的百分比,计算方法为定相的SNV的数量除以杂合SNV的数量。 |