摘要
- Lynch综合征相关PMS2基因的致病小变异检测会受到假基因PMS2CL的干扰。
- DRAGEN 4.3引入了一种改进的算法,可通过全基因组测序对PMS2基因中的小变异进行自主检测。
- 通过将该方法应用于22个非细胞系样本,检测到所有预期的P/LP变异。
- 这种方法可扩展到具有单拷贝或多拷贝旁系同源物的其他基因。
用于Lynch综合征筛查的PMS2致病变异检测会受到假基因PMS2CL的干扰
我们体内数万亿细胞中的每一个细胞都包含一套完整的DNA,而DNA本身又包含数十亿个亚基。如何在物理上创建和维护这一切呢?答案的一个重要部分是,我们的基因组包含有大量的纠错机制。其中一种机制称为错配修复(MMR),它可以识别并修复未正确配对的DNA。例如,如果A与G相对,而不是与T相对时,此机制就会启动修复。
如果一个人的MMR基因有缺陷,就会患上Lynch综合征,这是第二种最常见的癌症遗传易感性形式1。Lynch综合征的四个主要基因(MSH2、MSH6、MLH1和PMS2)中致病变异的总患病率在普通人群中可能高达1/2792。鉴于Lynch综合征患者中结直肠癌、子宫癌和其他形式恶性肿瘤的高发病率以及早期检测的重要性,准确的基因筛查在改善健康状况方面具有巨大的潜力。
由于假基因PMS2CL的存在,PMS2为Lynch综合征的基因筛查带来了特殊的挑战。这两个区域的高度相似性(图1)给变异检出带来了巨大难度3。源于该区域的read错配和模糊定位会导致假阳性或假阴性结果,从而影响临床决策和患者护理4。目前可用的方法可能需要专门的单基因湿实验室检测,如难以扩大规模的长片段PCR。
我们的目标是解决这些难题,利用因美纳流行的PCR-Free全基因组测序检测法提高PMS2第11-15号外显子小变异检出的可靠性。
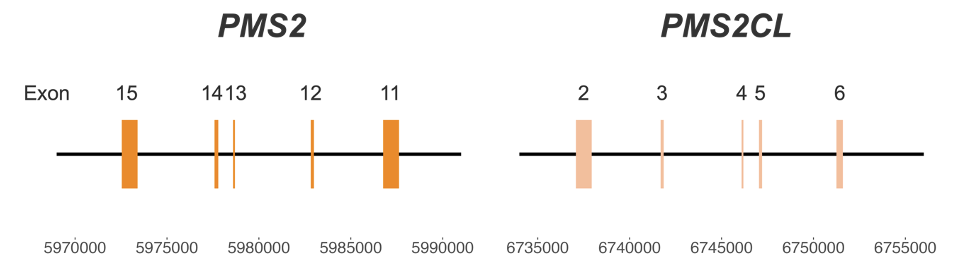
图1:PMS2和PMS2CL基因之间的高同源性区域——坐标来自hg38。
多区域联合检测方法
传统的变异检出程序依靠比对程序首先将序列read唯一地定位到其原始基因组位置,然后再检查是否存在微小差异。当read或read对只与参考基因组的一部分相似时,这种方法很有效,但当两个或更多区域匹配良好时,这种方法就很难奏效。由于至少5%的基因组有一个或多个近乎相同的拷贝,因此read的真正来源往往是不确定的。如果一组read的定位置信度较低,传统的变异检出程序可能会忽略这些read,即使它们包含有用的信息。如果read定位错误(即初步比对不是read的真正来源),就会导致变异检测错误。
为了应对这些挑战,我们开发了一种名为多区域联合检测(MRJD)的新型计算方法。MRJD并非孤立地考虑每个区域并单独对它们进行基因分型,而是考虑一组read可能从其中起源的所有位置,并尝试联合检测潜在的序列。这种方法可保留比对不明确的read,并适用于基因转换或交叉事件导致的read错误比对情况。
图2以PMS2和PMS2CL高同源性区域为例,展示了MRJD的一般工作流程。简而言之,MRJD在所有旁系同源区域中进行初步比对(无论定位质量如何),根据read与先验知识构建和定位所有单倍型,并计算联合基因型以检出小变异。
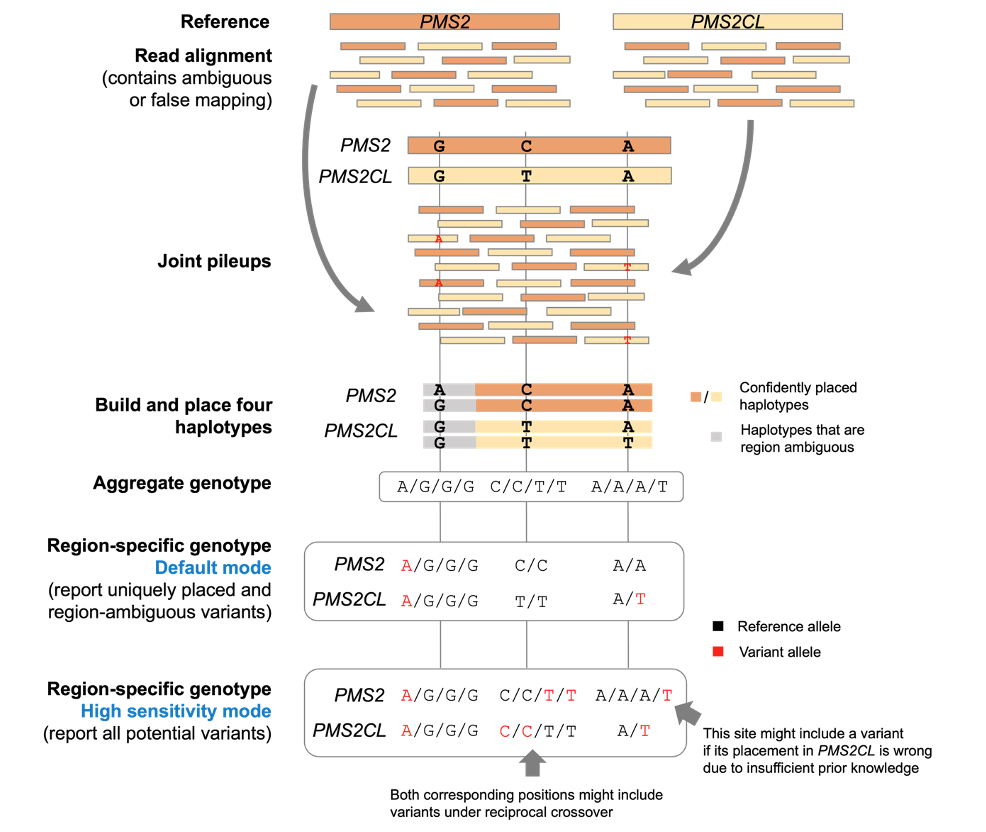
图2:MRJD工作流程。
对于灵敏度要求非常高的应用,建议使用MRJD高灵敏度模式
如图2所示,MRJD有两种模式:一种是在查准率和召回率之间取得平衡的默认模式,另一种是在牺牲查准率的情况下提供更大召回率的高灵敏度模式。在必须识别所有潜在致病变异,并可对这些变异的位置进行正交确认的情况下(如基于长片段PCR的检测),我们建议使用MRJD高灵敏度模式。
MRJD针对细胞系样本的性能
我们利用来自Illumina Polaris 1多样性panel的147个细胞系样本,对MRJD在PMS2高同源区域的变异检出性能进行了基准测试5。使用从长片段PCR数据中获取的正交小变异检出结果建立观测真值5。与DRAGEN小变异检出程序相比,MRJD的高灵敏度模式实现了更高的召回率,SNV和插入缺失的总召回率分别约为99.7%和97.1%(图3)。相比于长片段PCR数据中的SNV,插入缺失的召回率较低,这可能是由于插入缺失的错误率较高。为了解决这个问题,我们利用一个独立的数据集6,其中包括来自千人基因组计划的147个代表不同组源的细胞系样本,采用基于长读长的方法进行了一致性分析。相比基于长读长的方法,SNV和插入缺失的总召回率均高于99.7%。
MRJD高灵敏度模式的高召回率通过将所有可能的变异置于所有旁系同源区域中来实现,但其代价是查准率较低。为了衡量假检出率,我们将PMS2和PMS2CL中的合并正交变异检出与仅PMS2中的MRJD高灵敏度模式检出进行了比较。该分析表明,假检出率低于0.7%,这意味着几乎所有报告的等位基因都确实存在于样本中,但却被MRJD的高灵敏度(模糊定位)同时报告在两个位置上。
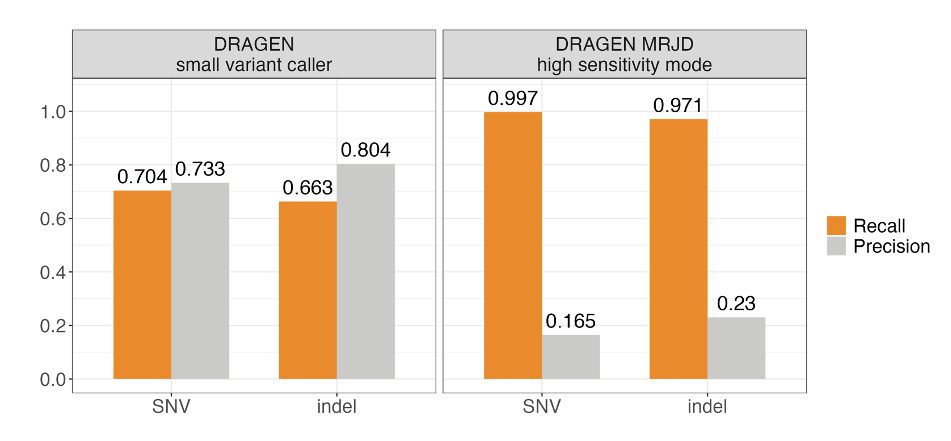
图3.DRAGEN小型变异检出程序(DRAGEN VC)和MRJD高灵敏度模式在来自Illumina Polaris 1多样性panel的147个样本上的总SNV和插入缺失性能——从长片段PCR数据中得出的正交小变异检出结果5。RTG Tools ploidy-squash模式用于生成基准统计数据。分析中排除了所有大于6 bp的完美同聚物和大于10 bp的不完美同聚物。
MRJD在非细胞系样本中的性能
为了评估MRJD方法在真实世界中的表现,我们与Broad Clinical Labs和Tempus AI合作,对总共22份非细胞系样本进行了MRJD性能评估,其中16份样本的PMS2和PMS2CL高同源性区域存在潜在的临床相关变异(11份来自Broad Clinical Labs,5份来自Tempus AI)。另外来自Broad的6份样本也被纳入了分析,这些样本具有与线状体肌病相关的已知致病变异,这些变异发生在NEB基因的三联区域,因为该区域是DRAGEN v4.3中MRJD所覆盖的另一个片段重复。
MRJD高灵敏度模式能够检测出所有22个样本中存在的所有预期临床相关小变异(表1)。
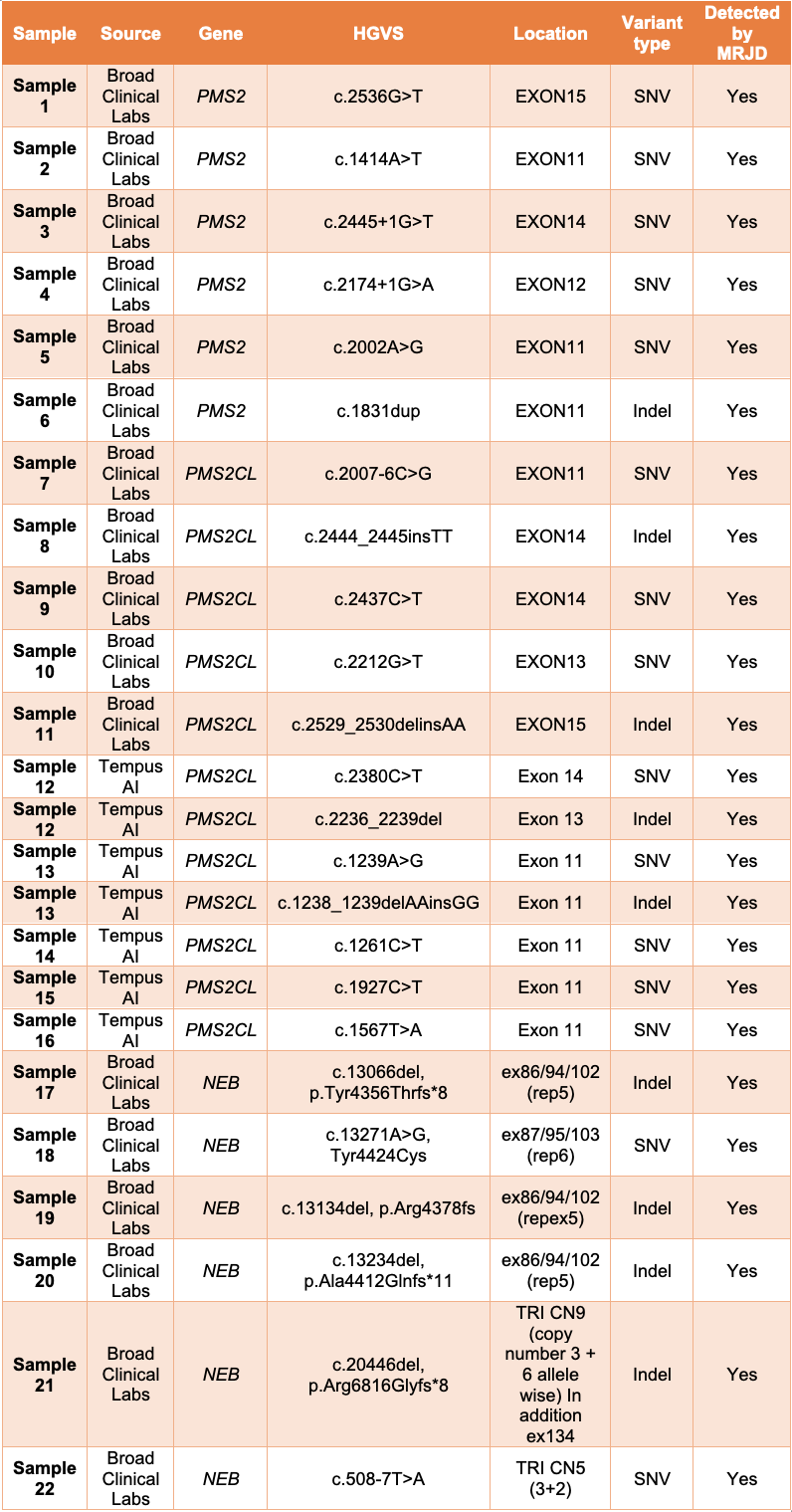
表1:22个非细胞系样本的MRJD高灵敏度模式结果,其中有潜在的临床相关变异——所有列出的变异都经过正交确认,并确定是PMS2、PMS2CL或NEB中的变异。结果由Broad Clinical Labs和Tempus AI提供。
估算假定未受影响样本中的P/LP变异检出率
高效的筛查和确认反复检测方法要求未受影响个体样本的总体阳性率较低。为了估算这一比率,我们使用来自千人基因组计划的147个细胞系测量了P/LP变异检出率。PMS2基因中的P/LP变异检出率为1/147(0.68%),表明未受影响个体样本中的反复检测负担较低。
讨论
总的来说,我们在此介绍了一种新颖的计算策略——多区域联合检测,它解决了利用WGS数据在旁系同源区域中进行从头生殖系小变异检出的难题,提高了灵敏度和特异性。通过将这种方法应用于PMS2和NEB基因,我们证实这项工作有助于更可靠地检测与Lynch综合征和线状体肌病相关的变异,从而更好地对受影响的个体进行风险评估并制定个性化的管理策略。
MRJD被设计为一个通用框架,适用于具有高序列一致性的旁系同源区域。除PMS2和NEB外,DRAGEN v4.3中的MRJD还支持其他五个临床相关和具有挑战性的基因重复区域中的生殖系小变异检出:SMN1、SMN2、STRC、IKBKG和TTN。据估计,人类基因组中有200-500个医学相关基因存在问题区域,其中高同源性是首要问题7-8。我们期望我们的方法可以为进一步研究面临类似同源性挑战的其他医学相关基因的变异检出铺平道路。
补充材料
除了本博客中介绍的结果外,我们还与Tempus AI合作,使用150个细胞系样本对MRJD检出程序进行了评估。这次合作的结果已在ISMB 2023会议上公布。有关详细见解,请点击此链接参阅相关出版物。值得注意的是,自本摘要发表以来,MRJD检出程序的性能有了进一步提高。
可用性
该软件在DRAGEN v4.3版本中提供,其安装文件和发行说明可点击此处.获取。有关MRJD检出程序的更多信息,请参阅此处的用户指南或联系ffg-info@illumina.com.
致谢
衷心感谢我们的合作伙伴——Broad Clinical Labs的Marina DiStefano和Edyta Malolepsza,以及Tempus AI的Francisco M. De La Vega和Pavana Anur,感谢他们采用非细胞系样本对MRJD检出程序进行的评估。
参考文献
- Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76(1):1-18. doi:10.1111/j.1399-0004.2009.01230.x
- Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer." Cancer Epidemiol Biomarkers Prev. 2017;26(3):404-412. doi:10.1158/1055-9965.EPI-16-0693
- van der Klift HM, Mensenkamp AR, Drost M, et al. Comprehensive Mutation Analysis of PMS2 in a Large Cohort of Probands Suspected of Lynch Syndrome or Constitutional Mismatch Repair Deficiency Syndrome. Hum Mutat. 2016;37(11):1162-1179. doi:10.1002/humu.23052
- Huang KL, Mashl RJ, Wu Y, et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell. 2018;173(2):355-370.e14. doi:10.1016/j.cell.2018.03.039
- Gould GM, Gauman PV, Theilmann MR, et al. Detecting clinically actionable variants in the 3′ exons of PMS2 via a reflex workflow based on equivalent hybrid capture of the gene and its pseudogene. BMC Med Genet. 2018;19(1):176. doi:10.1186/s12881-018-0691-9
- Chen X, Harting J, Farrow E, et al. Comprehensive SMN1 and SMN2 profiling for spinal muscular atrophy analysis using long-read PacBio HiFi sequencing. Am J Hum Genet. 2023;110(2):240-250. doi:10.1016/j.ajhg.2023.01.001
- Ebbert MTW, Jensen TD, Jansen-West K, et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 2019;20(1):97. doi:10.1186/s13059-019-1707-2
- Wagner J, Olson ND, Harris L, et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat Biotechnol. 2022;40(5):672-680. doi:10.1038/s41587-021-01158-1