最初记载
Guillaume Duchenne de Boulogne作为19世纪最具影响力的医学科学家之一值得人们铭记,他在整个职业生涯中为神经学做出了开创性的贡献。他是首批进行肌肉活检并将摄影成像应用于临床的科学家之一,并且发明了进行神经传导测试的设备。
他所照护的病人中,有一群患有严重肌肉萎缩的小男孩,他们的病症初期表现为行走困难,最终因膈肌功能衰竭,无法呼吸而死亡。直到1861年,Duchenne出版的书中讲述了他们的故事,这种病症才被大众知晓。现在这种病症以他的名字命名:杜氏肌营养不良症。
DMD相关病症的临床症状和自然病史
DMD是已知最大的人类基因之一,位于X染色体的区带Xp21.2-p21.1处。它编码抗肌萎缩蛋白,该蛋白是大型抗肌萎缩蛋白相关糖蛋白复合物的组成部分,在维持细胞骨架和细胞外基质之间的连接中发挥着关键作用,从而保护肌肉细胞膜的完整性1。
有三种不同的情况与DMD相关:
- DMD基因活性低或DMD基因失活的人患有杜氏肌营养不良症(DMD)。DMD是一种严重的、进行性的肌肉萎缩疾病。最早的症状是爬楼梯困难、步态蹒跚和频繁跌倒;患者在幼儿期就会出现这些症状。大多数患者在10-12岁左右开始依赖轮椅,并在20岁左右需要辅助呼吸。DMD患者的预期寿命为20-40岁2。
- 具有部分DMD基因活性的人患有贝克肌营养不良症(BMD)。BMD的特征是晚发性骨骼肌无力。发病年龄从5岁到60岁不等;与DMD相比,其病程更慢且更难以预测,预期寿命为40多岁3。
- 携带DMD致病变异的母亲有患DMD相关扩张性心肌病(DCM)的风险。该病症的特征是随着年龄的增长,运动功能减退、扩张性表型和充血性心力衰竭逐渐加重。
严重的DMD表型通常是由破坏阅读框的变异引起的,导致抗肌萎缩蛋白的表达完全丧失,而不太严重的BMD表型通常与框内突变有关,会产生异常但半功能性蛋白质(具有完整的N和C末端)4。
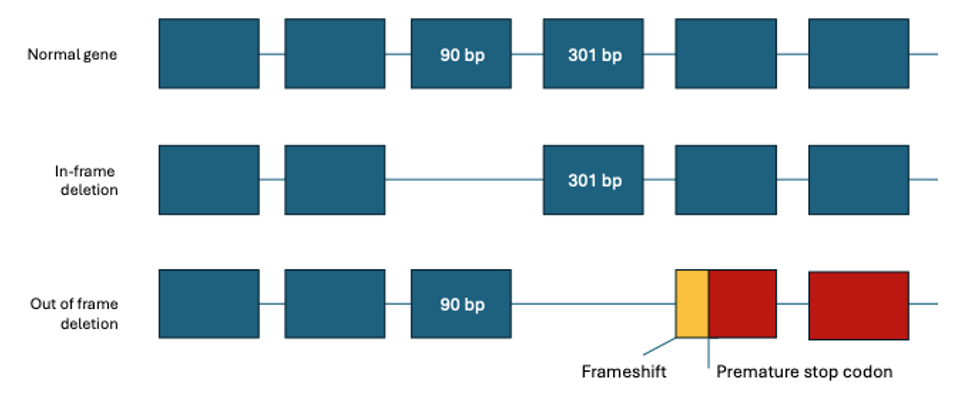
由于基因较大(基因组DNA超过2.2 Mb,包含79个外显子)且突变类型多样,突变分析历来具有挑战性。大多数已识别的突变为缺失类型,占60%-65%的DMD突变和85%的BMD突变,在大约5%-10%的突变中观察到重复。其余致病原因为小突变,例如插入/缺失,点突变或剪接突变6。据估计,约有25%-33%的病例是由新发突变事件引起的7。
导致杜氏肌营养不良症的原因是什么?
由于几乎所有的DMD病例都是男孩,并且同一家庭中可能有多人受到影响,但前提是他们有共同的母系亲属,因此最初推测DMD一定是由X染色体上的变异引起的。事实证明确实如此,该基因于1987年由Lou Kunkel及其合作者成功克隆8。这是早期疾病基因发现中的重要里程碑,发现时间介于HBB基因(镰状细胞性贫血,1985年)和CFTR基因(囊性纤维化,1989年)之间。
虽然单核苷酸变异和50 bp以下的小片段插入或缺失肯定会导致DMD,但DMD中80%的致病变异是一个或多个外显子的缺失或重复9。
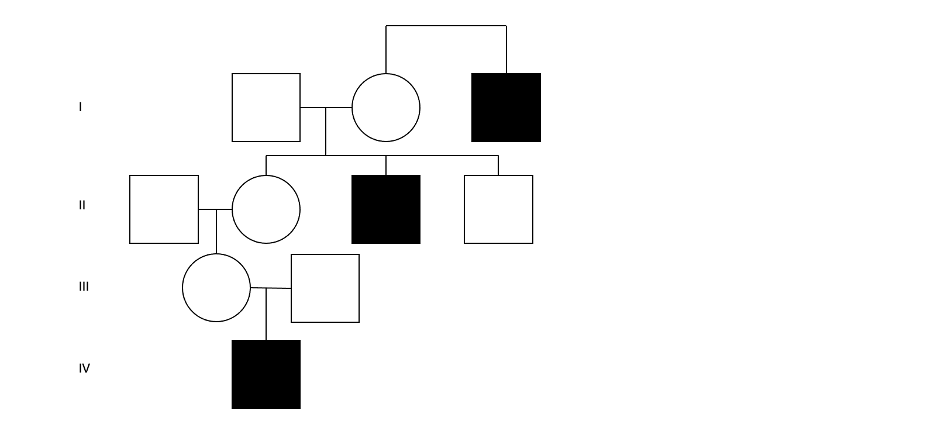
图2:假设四代人中有三人受DMD影响——这证明了X连锁隐性遗传的典型模式:多代受影响的男性共享母系血统。
DMD疗法
传统的对症干预措施,如支架和皮质类固醇,正开始被精准医疗方法所取代。对于特定外显子缺失的受影响个体,可使用反义寡核苷酸药物诱导跳过特定的剩余外显子,以恢复部分DMD基因功能10。这种方法高度依赖于准确的分子诊断。
2023年,一种更通用的基因疗法被批准用于DMD:Elevidys。该产品利用病毒载体将缩短版的DMD基因引入受体,使他们自身的肌肉细胞产生“微抗肌萎缩蛋白”。这种紧密的基因产品包含所有必要的结构和功能成分,美国食品药物管理局得出结论——其临床试验数据表明,这种效应“很可能预测年轻患者的临床获益”。这给针对此类疾病和其他疾病的基因治疗方法带来了希望。
DMD变异通常如何检测?
多重连接依赖性探针扩增(MLPA)是一种分析拷贝数变异(CNV)的常用技术,已广泛应用于DMD。这种基于扩增的方法可有效检测最常见的致病DMD变异,但有几个主要缺点,包括无法检测单核苷酸变异(SNV)和小片段插入缺失。
新一代测序(NGS)的最新进展和生物信息学算法的改进最终推动了基于NGS的单一检测方法的开发,用于检测所有变异类型,包括CNV和序列变异。全基因的全序列分析可以检测深层内含子致病变异,并准确检测涉及相似外显子的CNV断点,这可能会影响临床试验的结果11。
虽然所述的标准方法可以检测到影响编码区域的大多数变异,但亚外显子、亚内含子、深内含子或大型结构排列(例如倒位)可能无法检测到,因此有2%-7%的DMD病例仍未得到解决12。然而,临床全基因组测序可以捕获蛋白质编码和非蛋白质编码区域的大多数遗传变异,包括SNV、CNV和小结构变异(SV)。
基因组时代如何改变DMD的游戏规则?
全基因组测序(WGS)有望通过一种组合方法来解决这些限制,该方法可评估覆盖深度的变化并分析跨越断点的read。由于WGS在实验室之间和受试者之间提供一致性结果(与杂交捕获或基于扩增子的测序相反,后者在两方面的结果可能存在很大差异),因此任何人都可以使用为检测这些基因组重排事件而开发的方法。有关这些方法的更多信息,请参阅我们之前关于CNV-SV联合检出的博客文章。
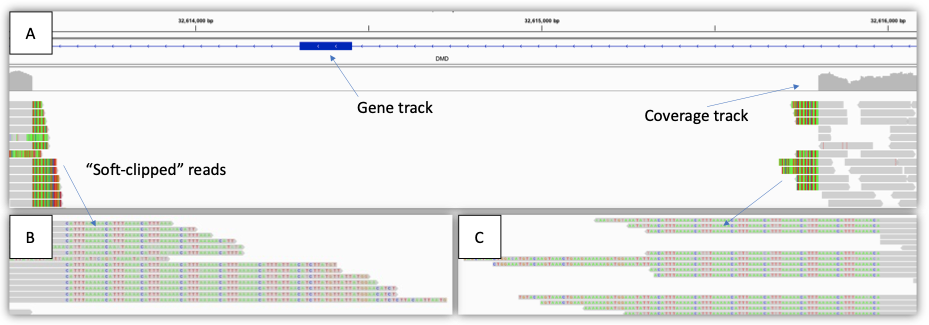
图3:杜氏肌营养不良症男性患者中单个外显子缺失(2 kb)的示例——(A) IGV视图显示受影响的区域,包括基因轨迹、覆盖轨迹和比对read轨迹。请注意,整个区域的覆盖度降至零,表明存在半合子缺失。(B)、(C) 放大的IGV视图显示“软剪辑”read。每个read的灰色部分与参考序列匹配,而彩色字母部分则不匹配(软剪辑)。在本例中,软剪辑read定位到缺失断点的另一端。这些read可用于生成SV检出。
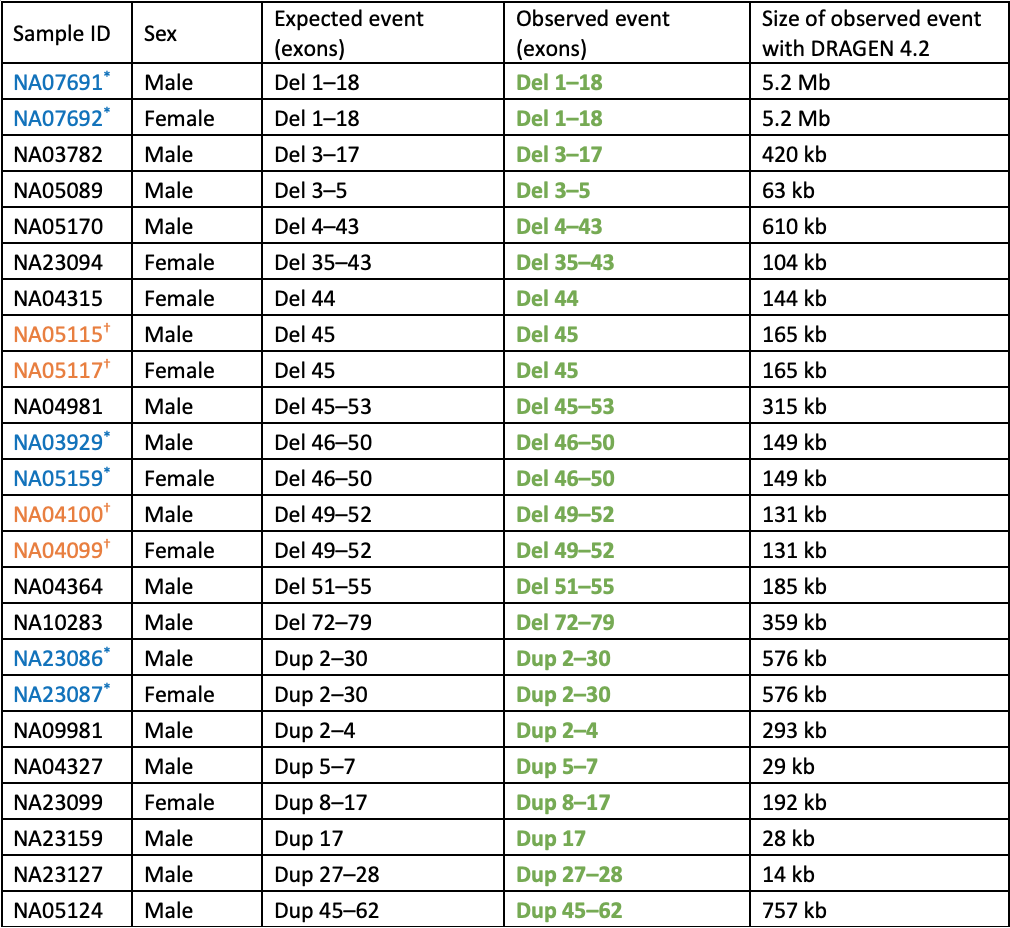
表1:通过WGS和DRAGEN 4.2联合SV检出程序分析具有已知DMD拷贝数变异的细胞系——所有预期事件均已检测到。样本ID以蓝色或橙色显示并带有*或†标志的连续条目为一对样本,其中男性是女性携带者的儿子。与预期外显子、CNV类型(del或dup)和接合性匹配的事件以粗体显示并设置为绿色。女性的所有事件都是杂合子,而男性是半合子。
值得注意的是,针对千人基因组计划数据集中剩余的约3200名个体,均没有检出假阳性外显子DMD拷贝数事件。这表明该变异类型的总体假阳性率非常低,这对于群体基因组学或携带者筛查研究等高通量项目至关重要。
结论
使用WGS和DRAGEN进行联合CNV和SV检出是一种极其灵敏的组合方法,能够检测出DMD中医学相关的缺失和重复。由于该基因可能是已知的CNV检出中最具挑战性的基因,尤其是在女性携带者中,因此证据表明WGS和DRAGEN 4.2设定了非常高的标准。这意味着您可以使用因美纳工具对DMD进行无缝研究评估,享受从样本到结果的一站式体验。
而且由于这是一种通用方法,因此其他具有医学相关性外显子水平CNV的基因也可以使用这些方法进行评估。具有检测重要性的类似变异示例包括LDLR(家族性高胆甾醇血症)、NF1(神经纤维瘤病)、BRCA1/2(遗传性乳腺癌和卵巢癌)和PRKN(早发性帕金森病)。
参考文献
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990 Nov;108(5):748-52. doi:10.1093/oxfordjournals.jbchem.a123276
- Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet. 2019 Nov 30;394(10213):2025-2038. doi:10.1016/S0140-6736(19)32910-1
- Darras BT, Urion DK, Ghosh PS, et al. Dystrophinopathies. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews. 2000 Sep 5. PMID:20301298
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1998 Jan;2(1):90-5. doi:10.1016/0888-7543(88)90113-9
- Le Rumeur E. Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies. Bosn J Basic Med Sci. 2015 Jul 20;15(3):14-20. doi:10.17305/bjbms.2015.636
- Ji X, Zhang J, Xu Y, et al. MLPA Application in Clinical Diagnosis of DMD/BMD in Shanghai. J Clin Lab Anal. 2015;29(5):405-411. doi:10.1002/jcla.21787
- Santos R, Gonçalves A, Oliveira J, et al. New variants, challenges and pitfalls in DMD genotyping: implications in diagnosis, prognosis and therapy. J Hum Genet. 2014;59(8):454-464. doi:10.1038/jhg.2014.54
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509-517. doi:10.1016/0092-8674(87)90504-6
- Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395-402. doi:10.1002/humu.22758
- Matsuo M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021;4(3):232-240. doi:10.31662/jmaj.2021-0019
- Nallamilli BRR, Chaubey A, Valencia CA, et al. A single NGS-based assay covering the entire genomic sequence of the DMD gene facilitates diagnostic and newborn screening confirmatory testing. Hum Mutat. 2021;42(5):626-638. doi:10.1002/humu.24191
- Andrews JG, Galindo MK, Thomas S, Mathews KD, Whitehead N. DMD Gene and Dystrophinopathy Phenotypes Associated With Mutations: A Systematic Review for Clinicians. J Clin Neuromuscul Dis. 2023;24(4):171-187. doi:10.1097/CND.0000000000000436