简介
随着越来越多的实验室将全基因组测序用于临床检测,端到端测序流程性能的准确测定变得越来越重要。方法之一是获取参考材料并对其进行测序,然后根据相关真实数据来衡量变异检出性能1-3。借助这种方法,实验室可以精确测量基因组中大部分区域(80%-90%)的召回率和精确率。然而,这些参考材料局限于有限数量的种族同源样本,并不一定能预测未来样本的全基因组表现。一种互补的方法就是开发一种预测方法来估计任何样本的测序性能。
随机变异和系统变异都会影响到变异检出性能。对于单核苷酸变异(SNV)和短插入缺失等小变异来说,随机变异对大部分覆盖度≥30×的基因组的变异检出性能的影响非常小。但是,对于因低质量而受到系统性影响的基因组区域,如错误率升高、定位质量低或测序深度异常,即使是SNV和插入缺失,也可能无法提供持续准确的变异检出。有许多众所周知的系统特征可能会导致这些系统误差,例如,高度重复区域的定位质量较差,同聚物导致碱基准确性较低。我们利用这些知识将基因组划分为“简单”和“困难”区域4。虽然这些分类很有帮助,但它们并不能完全代表这些区域内的实际表现。例如,一个大的片段重复可能由高相似性和低相似性区域组成,从而导致变异检出准确度大相径庭。与这些通用分类方法相比,一种改进方法是使用实际测序数据,根据经验确定高变异检出性能和低变异检出性能的区域。
在此,我们分析了对29个样本进行测序(平均深度大于50×)后收集到的与数据质量相关的几项测序指标。合并后,我们平均有1,450个read覆盖基因组中的每个碱基,这让我们能够以碱基级分辨率稳定地识别出高质量的区域,并根据经验将每个位置注释为具有高或低的系统质量。然后,我们通过成对SNV的一致性进行评价,从而证明这些注释对SNV性能有很高的预测性。借助这些根据经验得出的注释,我们可以在常用的基于参考分类的方法上进行改进。例如,我们注释为高置信度的区域位于Genome in a Bottle(GIAB)困难区域内,在技术平行重复中具有较高的SNV一致性(Jaccard指数=98.8%)。相反,我们注释为低置信度的不在GIAB困难区域内的区域,其SNV一致性较低(Jaccard指数=79.9%)。我们的研究结果表明,通过汇总来自许多样本的序列数据,我们可以确定小变异检出系统性质量较高的基因组区域。
方法
为了建立数据库,我们从Coriell医学研究所生物库中选取了29个样本,这些样本已纳入千人基因组计划5。这些样本代表了不同的种族(11个非洲样本、9个东亚样本和9个欧洲样本)。使用TruSeq™ PCR-Free Sample Prep Kit制备每个样本的DNA,然后在NovaSeq™ 6000仪器上使用Xp工作流程进行双端150 bp read测序,平均深度为51×。
使用Dragen™ v3.4.5将序列read与带有decoy染色体但无替代contig的GRCh38进行比对。由于我们有29个样本,因此预计基因组中每个位置大约有1,450个read。我们从基因组中每个位置的所有样本的比对文件(BAM或CRAM)中收集了几个指标,包括:
- 归一化深度: 对于每个样本,归一化深度是通过将每个位置的覆盖度除以常染色体覆盖度的中位数来计算的,因此值1对应于常染色体覆盖深度的中位数。计算所有样本的归一化深度的平均值,得出最终的队列深度。
- 平均定位质量: 与基因组位置重叠的所有样本中所有配对read的平均定位质量。
- %Q20碱基检出: 所有样本中某个基因组位置的Phred标度质量评分超过20分(对应错误率≤1%)的碱基检出百分比。
在本分析中,我们使用通过Jaccard指数计算的一致性来衡量SNV的性能:
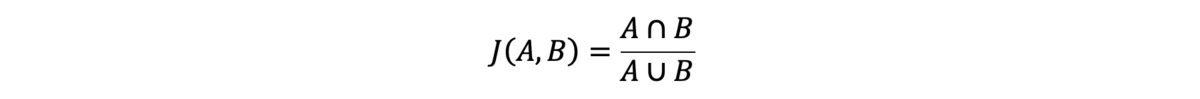
A和B是来自两个变异检出文件(VCF)的两组变异检出,通常来自同一样本的重复分析。 在分子中,A和B的交集定义为在具有相同基因型的两个VCF中存在的所有变异检出。 在分母中,A和B的并集定义为任一VCF中存在的所有变异检出。 这些变异检出集合的Jaccard指数的计算方法是,将这些集合中的检出集合的交集除以它们的检出集合的并集。
与真值资源相比,一些可重现的变异可能是由于系统误差造成的,但其结果对于一般性能评估仍具有很高的参考价值。在所有我们的分析中,我们显示了使用了NA12878的30个随机组合技术重复的Jaccard指数平均值。报告的每类变异的一致性值和变异数量是所有30个重复的平均值。
在计算常染色体性能时,我们排除了参考基因组中的N和缺口。
结果
汇总数据非常有助于检测在单个测序实验中可能无法检测到的系统误差趋势。在常染色体覆盖度中位数为30×的基因组中,考虑一个有40条read的位点。如果我们假设泊松分布为30×,那么在每1,000个独立位点中,大约有14个位点会出现这种情况。因此,很难确定观测到的较高覆盖度是随机的还是系统的。反之,在观察一个有30个个体、覆盖度为30×的群体中的同一位点时,如果每个个体在该位点的覆盖度都正好为40×,我们就可以非常确定(p=2e-56),该位点的覆盖度变化不是由随机过程驱动的。通过汇总从许多样本中获得的高深度数据,我们能够识别最有可能导致变异检出性能不佳的系统性异常。在来自不同种族背景的大量样本中,常见的突变和等位基因特异性杂峰是可以识别的,如果足够罕见,那么这些杂峰对汇总指标的影响将是有限的。
根据指标性能对基因组进行分层
我们按照“方法”中概述的程序收集了三个性能指标(图1)。
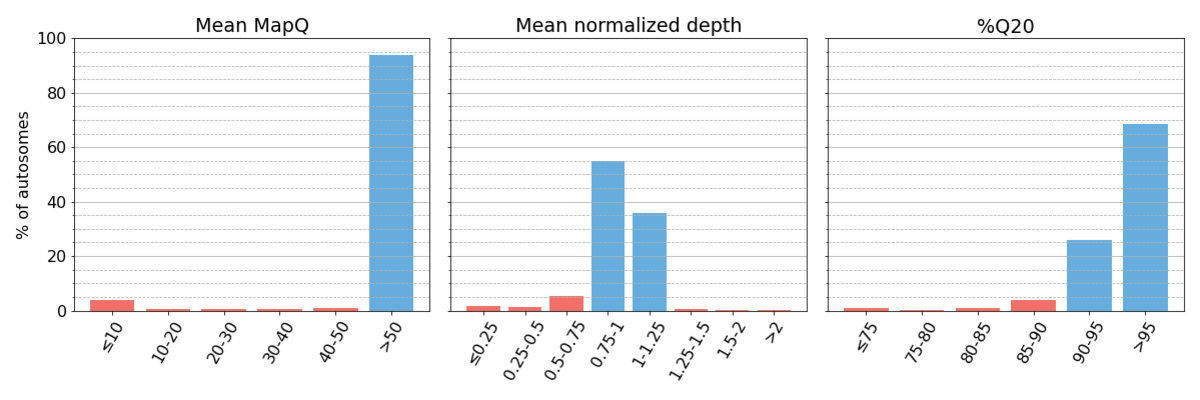
图1: 汇总测序数据的全基因组区间分类系统校对指标。指标区间为半开放状态。代表高系统质量的区间用蓝色阴影表示,代表低系统质量的区间用红色阴影表示。文章中的所有图表均采用这种方法。如文中所述,当区域的Q20≥90%、平均定位质量≥50、平均归一化深度在目标覆盖范围的25%以内时,我们将其定义为具有较高系统质量的区域。
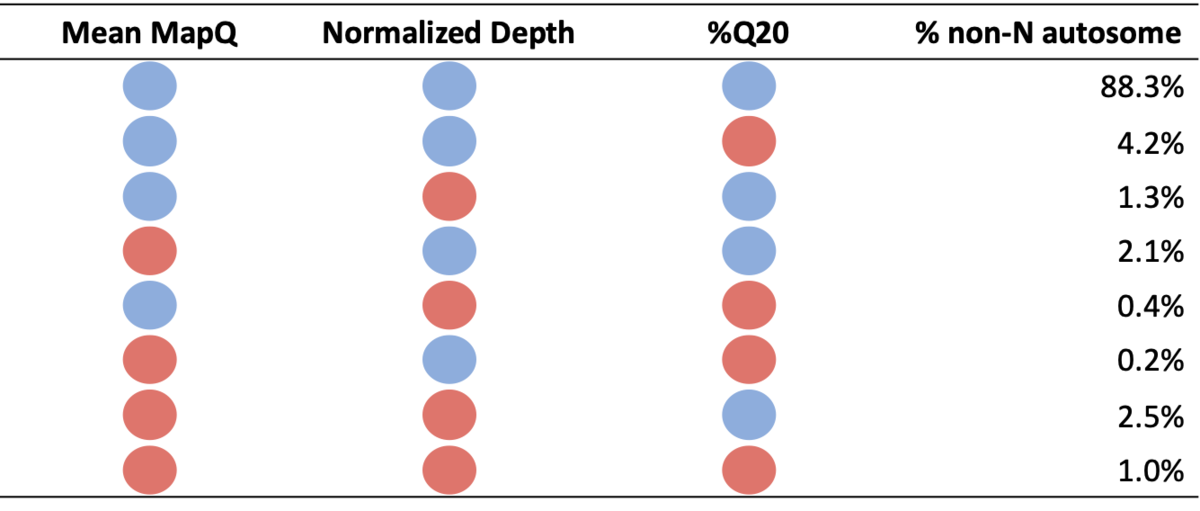
表1:非N常染色体在各系统质量分区中的百分比。蓝色圆圈表示特定指标的系统质量高,红色圆圈表示系统质量低。
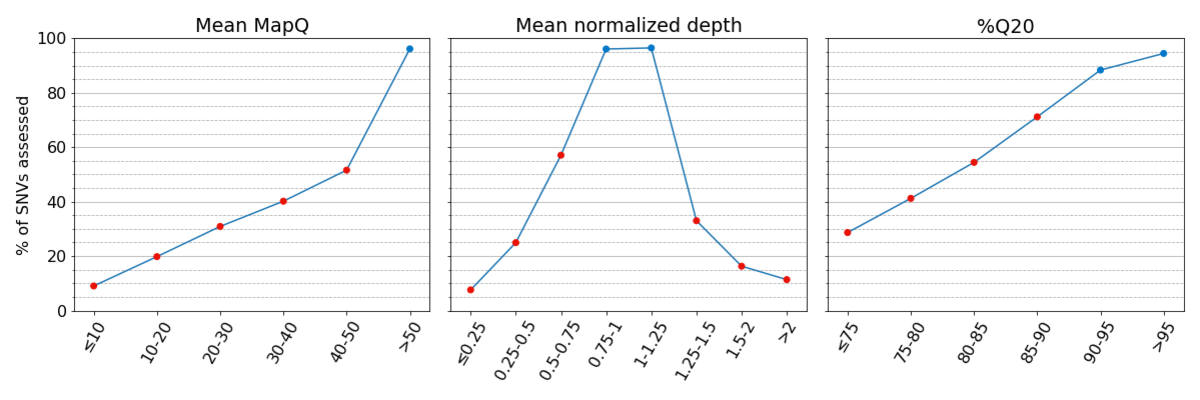
图2:在系统质量较低的区域,真值集的可评估SNV很少。基于Platinum Genomes v2017.1真值集给出了每个分区的已评估SNV百分比
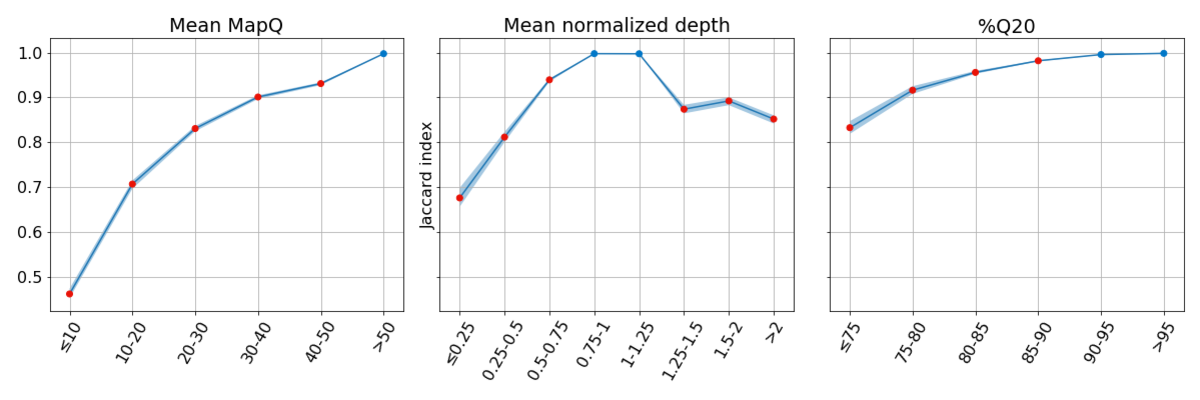
图3:在系统质量较低的基因组区域,一致性较差。在每个分区中,每个点的一致性基于其他两个指标具有较高系统质量时被得出,突出了低系统质量对每个指标单独的影响。
将经验系统质量与Genome in a Bottle困难区域进行比较
虽然基因组的参考特征会对变异检出性能产生负面影响,但这些区域的变异检出准确率并不明确。仅根据参考特征排除变异可能会排除许多高质量的变异检出。例如,Genome in a Bottle 定义了几类基因组的困难区域, ,包括低定位能力、片段重复、长串联重复以及GC含量极高的区域(4)。在每个类别中,18%-73%的困难区域在本分析中标记为高系统质量(表2)。对于属于“困难”类别的约61.8万个SNV,一致性为98.8%,在本分析中视为具有较高的系统质量。此外,在约11.8万个根据GIAB定义不属于困难区域的SNV中,经过我们的分析,认为其系统质量较低,一致性为79.9%。综合来看,这些结果表明,之前描述的困难区域过于宽泛,使用经验系统质量可以更好地预测生殖系变异检出性能。
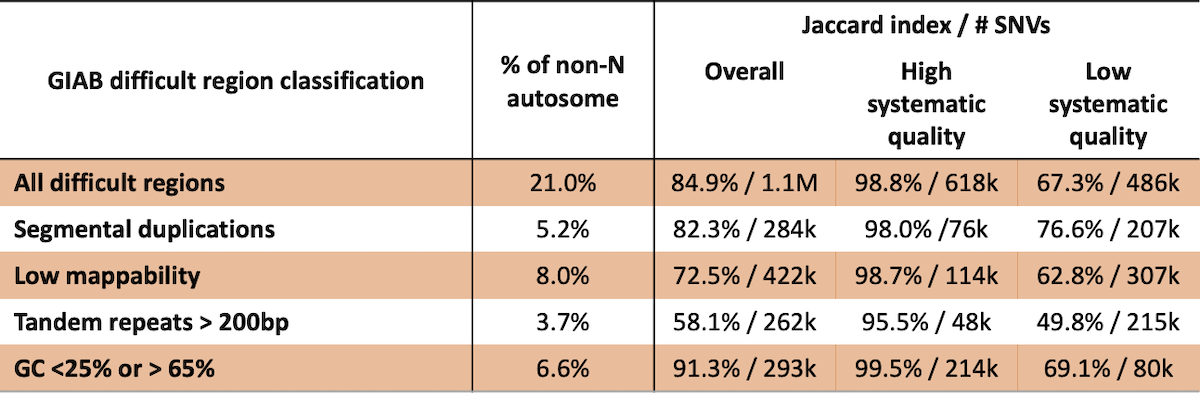
表2:GIAB困难区域对于变异检出性能的预测效果较差。
将经验系统质量与真值集置信区域进行比较
真值集置信区域特征产生的高置信度变异检出区域应与系统质量分区产生的区域相似,但不完全相同。此外,即使是同一样本,为这些真值集确定的置信区域也会随着时间的推移而变化。为了研究这一点,我们根据不同真值集置信区域与根据经验定义的置信区域的重叠程度,比较了不同真值集置信区域的性能(表3)。特别重要的是,既在真值集置信区域内又具有较高系统质量的位点具有一致的高一致性(>99.9%)。同样,不在真值集置信区域内且系统质量较差的位点的一致性也很差。
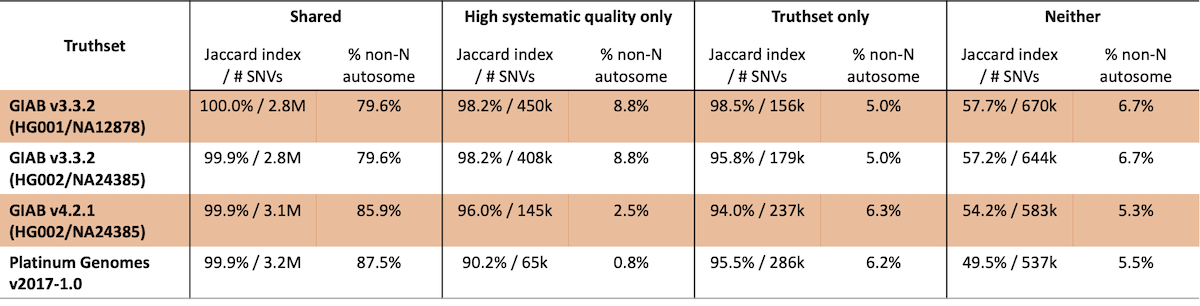
表3:与高系统质量区域相比,真值集的SNV一致性
在这项分析中,发现了6.5万到45万个SNV,这些SNV在所研究的真值集中并不存在,一致性范围为约91%到>98%,表明这些位点总体上质量较高。 这些观察结果也印证了本分析的粗略程度——随着方法的发展,我们希望高系统质量位点的一致性能进一步提高。
根据这些观察结果,我们得出了两个进一步的结论。首先,鉴于此处考虑到了分区的粗略性,因此,通过更复杂的分析,这些真值集特异性变异中的某些部分最终可能会归类为具有高系统质量。其余12%基因组中的某些子集能够产生变异检出,这些检出需要正交验证才能视为具有高置信度。这正是真值集所能提供的 — 无论是通过使用谱系一致性、正交平台和流程,还是手工整理,这些额外的证据都能在基因组更困难的区域实现高质量的检出。
讨论
为了成功推动基因组数据临床应用的改进,关键是要全面了解我们的性能,不仅是在广泛的全基因组范围内,还有在高度局部范围内。通过汇总大量人群的全基因组数据,我们可以开始获取关于任何目标区域性能的详细信息。在观察到对重要基因的特定影响时,我们可以根据这些信息开发专门的检出程序,利用NGS中的可用信息提供临床上可操作的结果,因美纳为SMA和CYP2D6定制的检出程序就证明了这一点6,7。通过细分我们的小变异检出性能,我们对系统误差如何影响特定应用性能有了越来越清晰的认识。这对今后的应用起到指导作用,我们可以借此了解目前可以获得可靠临床结果的具体方面,并指出了临床基因组分析改进的下一个关注重点。
本文介绍的结果表明,我们有望开发一套通用的置信区域,这套区域的重点是能预测变异检出性能的特征。我们证明,在高系统质量区域内,通过成对重复一致性评估的SNV性能非常高。重要的是,由于高系统质量区域是在相对于评估集的不同样本上定义的,因此这些结果很可能适用于任何测序基因组。目前,这项工作发现,在大于88%的常染色体中,SNV检出的重复率约为99.8%,这表明基因组的这一部分几乎不需要正交验证。研究人员目前正在继续开展工作,以更好地确定覆盖基因组更大比例的高置信度区域。此外,这项工作主要集中在SNV一致性上,但这些方法同样可以推广到更复杂的变异类型以及体细胞变异检出。
参考文献
- Eberle MA, Fritzilas E, et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017;27(1):157-164. doi:10.1101/gr.210500.116
- Wagner J, Olson ND, et al. Benchmarking challenging small variants with linked and long reads. bioRxiv. 2020;212712. doi:10.1101/2020.07.24.212712
- Zook JM, McDaniel J, et al. An open resource for accurately benchmarking small variant and reference calls. Nat Biotechnol. 2019;37:561-566. doi:10.1038/s41587-019-0074-6
- Krusche P, Trigg L, et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat Biotechnol. 2019;37:555-560. doi:10.1038/s41587-019-0054-x
- Clarke L, Fairley S, et al. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017;45(D1):D854-D859. doi:10.1093/nar/gkw829
- Chen X, Sanchis-Juan A, et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet Med. 2020;22:945-953. doi:10.1038/s41436-020-0754-0
- Chen X, Shen F, et al. Cyrius: accurate CYP2D6 genotyping using whole-genome sequencing data. Pharmacogenomics J. 2021;21:251-261. doi:10.1038/s41397-020-00205-5