简介
新一代测序(NGS)助力科学家破译基因组,更深入地了解生物学。因美纳成熟的边合成边测序(SBS)化学技术结合屡获殊荣的DRAGEN二级分析,可提供高准确度的全基因组测序(WGS)数据1,2。 DRAGEN多基因组(Graph)进一步提升了复杂区域的定位准确度,提高了大约50%1。然而,仍然存在一小部分基因区域,仅使用短读长难以绘制,却可以得益于较长的读长,从而提高定位能力。
Illumina Complete Long Reads提供一体化的工作流程,实现了长读长测序的可及性,并有助于分析人类基因组中的复杂区域。Illumina Complete Long Reads使您在同一平台上就能完成长读长和短读长测序。结合DRAGEN信息学和机器学习方法,Illumina Complete Long Reads可以通过NGS技术提取准确的变异检出和相位信息。本文深入探讨了Illumina Complete Long Read人类基因组分析背后的基本原理。
工作原理:检测概览
Illumina Complete Long Reads工作流程(图1)结合了专利的文库制备分析、成熟的因美纳SBS化学技术和强大的DRAGEN二级分析,可生成高度准确的长读长数据,N50为5–7 kb。
Illumina Complete Long Reads的文库制备
高效的文库制备方案只需一天即可完成,并且易于扩展,适用于高通量研究,DNA起始量仅需50 ng。*这种检测使用转座酶片段化生成基因组DNA长片段(>10 kb),无需额外的剪切或片段长度筛选。单分子DNA长片段以独特的单碱基对改变模式被酶标记,这些“界标”被低频(4%-7%)引入到DNA片段的各个位置。每个单分子片段都具有独特的界标标记,以捕获和保存长读长信息(无需使用复杂的条形码或接头)。界标标记的长片段首先会被扩增,然后经过第二次转座酶片段化步骤进行文库制备,以便在因美纳系统上进行标准测序。
* 建议的DNA起始量为50 ng,可低至10 ng。
生物信息学工作流程
分析流程中生成的长读长数据与未标记的标准WGS文库相结合†,生成连续的长读长,可完整、准确地展示原始单分子片段。
† 需要来自同一样本的30×标准短读长人类全基因组数据用于分析。推荐使用Illumina DNA PCR-Free Prep,也兼容第三方WGS试剂盒。未标记的文库无需同时制备或测序,可以使用之前运行的样本中的FASTQ文件。
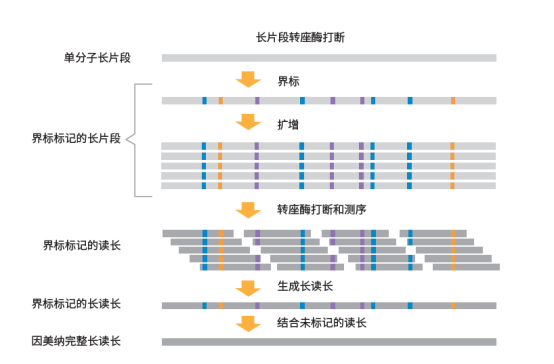
这种检测使用转座酶片段化生成基因组DNA长片段(>10 kb),无需额外的剪切或片段长度筛选。每个单分子片段都具有独特的界标标记,以捕获和保存长读长信息。界标标记的长片段首先会被扩增,然后经过第二次转座酶片段化步骤进行文库制备和测序。分析流程中生成的长读长数据与未标记的标准WGS文库相结合(来自于同一样本,单独分析),去除界标,产生高准确度的完整长度长序列。
> 观看工作原理
Illumina Complete Long Reads生成
Illumina Complete Long Reads长读长生成的生物信息学工作流程包括标准基因组计算方法,如比对与变异检出。该工作流程被包装成BaseSpace Sequence Hub中的一键式应用软件,并使用界标标记和未标记的文库以及参考基因组作为起始量。这些起始量随后被用于执行一系列步骤(图2),从单分子中生成长读长,实现全面的WGS分析。

识别片段中的界标标记位点
长读长生成过程的第一步是识别界标标记文库中存在的标记。在可信绘制区域中,大多数界标标记可以通过标准比对和检测与参考基因组不同的核苷酸来识别。
如果片段来自难以与参考基因组比对的区域(例如,重复区域),则需要采用不同的方法来检测界标标记。针对k-mer(即,将片段信息分解成“k”长度的小核苷酸串)的特定方法让算法能够在不使用参考基因组的情况下确定片段之间的关系。在难绘制区域中,标记位点的推断通过比较界标标记与未标记片段中的k-mer进行3。如果标记片段中的k-mer与未标记片段中的任何k-mer均无法匹配,则该k-mer将被视为界标标记。
构建界标标记片段的加权网络
在检测出片段中的所有界标后,下一步就可以根据它们的共用标记来识别片段之间的连接。我们使用minimizer k-mer来索引相似的片段对,并优化k-mer匹配4,这种方法可以详细比较所有共用给定minimizer k-mer的片段对。共用界标和冲突界标的数量决定了片段之间的连接强度(图3)。我们根据这些连接强度构建了标记片段的加权网落。

共用界标的片段连接到同一个网中,连接强度取决于共用界标和冲突界标的数量。在右图中,较强的连接用加粗的线条表示,较弱的连接用虚线表示。
寻找来自相同模板的片段组
各片段之间的连接使所有片段形成一张图,应用一系列分解和成簇方法(例如,移除共用界标数量少的冲突或弱连接),将整个网拆分成紧密连接的簇(图4)。假定每个簇都来自单个分子。

连接图按照最强连接拆分。根据推断,每个紧密连接的簇由来自同一模板分子的片段组成。
组装各个界标标记的片段组
在最终的簇中,DRAGEN分析使用基于k-mer的de Bruijn图组装法生成长读长的重叠群(图5)。

将每个簇(对应一组据推断来自同一模板分子的片段)组装进界标标记的长读长。
去除长读长中的界标
在将界标用于支持长读长的生成后,可去除界标。为了区分界标和真实变异,将界标标记的长读长与未标记的片段比较。更新任何与相应的未标记片段不匹配的界标,使最终的Illumina Complete Long Read显示真实序列(图6)。比较界标标记的长读长与未标记片段的方法类似于界标的识别方式:部分使用参考基因组比对,部分使用k-mer索引,特别是在难绘制区域。在获得未标记片段与界标标记的长读长的比对结果后,应用贝叶斯模型确定长读长的最终碱基识别和相应的质量分值。

将每个组装得到的界标标记的长读长与未标记片段比较,区分界标(方块)和真实变异(圆圈)。更新与未标记片段冲突的界标标记的碱基,使之匹配未标记片段。最终的Illumina Complete Long Reads准确地展示原始单分子并显示真实序列。
二级分析
完成上述的Illumina Complete Long Read构建步骤之后,使用Illumina Complete Long Reads和未标记的短读长执行二级分析(图7)。首先,使用改进版本的Minimap2将完整长读长与基因组比对。
对于小变异检出,将来自长读长和短读长的DRAGEN小变异检出结果合并为同一个VCF文件。DRAGEN小变异检出能够处理长于75 kb的片段。使用机器学习模型(在“瓶中基因组”的变异检出中训练)来合并和改进从长读长和标准短读长中获得的小变异检出。最后,使用改进版本的WhatsHap定相Illumina Complete Long Reads和合并的小变异,并创建新的全面输出文件以捕获单倍型信息。
对于结构变异检出,将长读长结构变异检出程序(Sniffles2)输出5和短读长DRAGEN结构变异检出程序输出合并为同一个VCF文件。

(A)分别比对长读长和短读长,然后将结果与高级逻辑结合,从而优化变异检出。使用定相工具定相长读长和合并的小变异。(B)使用专用的SV检出程序分别对长读长和短读长执行结构变异(SV)检出,将结果用高级逻辑合并,创建新的、合并的SV VCF文件。
高度准确的WGS
llumina Complete Long Read技术利用了成熟的因美纳SBS化学技术和DRAGEN二级分析,进一步提高人类全基因组测序的准确性。PrecisionFDA Truth Challenge v2数据集显示,使用Illumina Complete Long Read检测的F1评分为99.87%,F1评分反映WGS的精确率和召回率(图8)6,7。与标准WGS相比,Illumina Complete Long Read数据显示,在多个基准样本中的SNP和插入/缺失中假阴性和假阳性全面减少(图9)。

PrecisionFDA Truth Challenge v2数据集显示,Illumina Complete Long Read Prep,Human(橙色)提供了高度准确的变异检出(以F1评分(%)衡量,该评分反映WGS的精确率和召回率)。使用Illumina DNA PCR-Free Prep和DRAGEN 4.0(黄色)或其他市售的长读长解决方案(紫色)的标准WGS无法达到这种准确度。

以瓶中基因组联盟人类参考样本HG002、HG003和HG004,测定单核苷酸多态性(SNP)和插入/缺失变异检出准确性的假阳性(FP)和假阴性(FN)。比较来自Illumina Complete Long Read(橙色)和Illumina DNA PCR-Free Prep(黄色)的WGS数据,这些数据涉及全基因组。
结论
长读长信息有助于分析复杂基因组区域。Illumina Complete Long Reads支持在同一台仪器上进行长读长和短读长测序,助力基因组学实验室轻松实现全面的WGS。Illumina Complete Long Reads具有许多优势,例如熟悉的一体化实验室工作流程、起始量要求低、大规模的文库试剂盒制造以及连续的读长,从而产生高质量和全面的基因区域变异检出。
了解更多
Illumina Complete Long Reads产品线
阅读有关使用Illumina Complete Long Reads提高小变异检出准确性的方法的信息:使用Illumina Complete Long Read Prep, Human进行全面的全基因组测序
Illumina Complete Long Read Prep,Human数据表
Illumina Complete Long Reads技术
参考文献
- Mehio R, Ruehle M, Catreux S, et al. DRAGEN Wins at Precision- FDA Truth Challenge V2 Showcase Accuracy Gains from Alt-aware Mapping and Graph Reference Genomes. Accessed May 16, 2023.
- Illumina. Accuracy improvements in germline small variant calling with the DRAGEN Bio-IT Platform. Accessed May 16, 2023.
- Leinonen M, Salmela L. Extraction of long k-mers using spaced seeds. IEEE/ACM Trans Compu Biol Bioinform. 2022;19(6):3444-3455. Doi:10.1109/TCBB.2021.3113131
- Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA. Reducing storage requirements for biological sequence comparison. Bioinformatics. 2004;20(18):3363-3369. doi:10.1093/bioinformatics/bth408
- Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15(6):461-468. doi:10.1038/s41592-018-0001-7
- Illumina. Data on file. 2022.
- PrecisionFDA. Truth Challenge V2: Calling Variants from Short and Long Reads in Difficult-to-Map Regions. precision.fda.gov/ challenges/10. Accessed January 12, 2023.