
因美纳与阿斯利康开展战略研究合作,以加速药物靶点发现
将结合双方在基于人工智能(AI)的基因组解读和基因组分析技术优势及行业专长,加速药物靶点的发现...
因美纳药物开发解决方案

DiscovEHR研究中50,726个全外显子组序列中的功能变体分布及其临床影响
近年来,大规模遗传学研究揭示了人类基因组中的罕见和常见变异,为探索新的生物学机制和治疗靶点提供了重要线索。精准医疗的实施依赖于对这些遗传因素的深入研究。为此再生元遗传学研究中心(Regeneron Genetics Center)和格伊辛格卫生系统(Geisinger Health System)联合对超5万名美国人进行相关研究,并将研究结果《Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study》发表在了世界级顶刊Science(IF=44.7)上。该研究通过整合参与者的高通量外显子组测序结果和纵向电子健康记录(EHR),寻求功能变体与临床表型的关联。

研究背景
在人类遗传学研究领域中,通过大规模的基因组测序技术,可以揭示许多未知的基因变体及其生物学功能,从而为疾病的机制研究和新药的靶点发现提供重要线索。特别是在集成健康系统中,这些研究能够充分利用电子健康记录(EHR)数据,推动精准医疗的发展。为此,Regeneron Genetics Center与Geisinger Health System合作启动了DiscovEHR项目,通过高通量外显子组测序,对基因进行详尽分析,并将基因数据与2014年的EHR数据相整合。
研究涉及的参与者主要为欧洲血统,通过分析这些参与者的外显子组数据,研究者试图了解罕见的基因功能丧失(LoF)变体的分布及其临床影响。尽管这些变异通常发生频率极低,但由于其可能导致基因功能的丧失,因此通过对大规模数据的对比分析,仍能帮助研究者发现相关疾病的遗传学基础。
样本来源及方法
1. 样本采集与参与者选择
研究对象来自Geisinger Health System的“我的健康码社区健康计划”,涵盖了50,726名成年参与者。这些参与者主要为欧洲血统,并同意提供血液和DNA样本用于广泛的研究目的,包括基因组分析和连接电子健康记录(EHR)的数据。
2. 高通量全外显子组测序
利用Illumina HiSeqTM 2500平台(仅供科研使用,不得用于诊断),研究团队对参与者的DNA样本进行了高通量外显子组测序,旨在覆盖18,852个基因的编码区域。每个样本的读深度足以确保至少85%的目标区域达到20×的单倍体读深。
3. 数据处理与变异识别
通过软件将测序结果与标准基因组参考进行对比,并完成基因型分配、变异注释和功能预测。此外,还使用了GATK VQSR质控方案对测序结果进行数据质量评估与控制,将低质量测序数据排除。
4. 关联分析与验证
利用EHR数据,对参与者的临床表型进行系统化分析。通过混合线性模型,进行基因负担测试和外显子组关联分析,并将模型中的变异与ClinVar和HGMD等数据库进行比对,确定其是否为已知的致病变体,从而寻找出基因变体与临床表型(如血液学特性等)之间的关联。
结果
表1 DiscovEHR研究对象(≥18岁)的人口学和临床信息统计表

表1提供了一个全面的参与者人口学和临床特征的概览,显示了DiscovEHR项目参与者的多样性和健康状况。
表2 在50,726名DiscovEHR参与者中通过全外显子组测序鉴定出的序列变体统计表
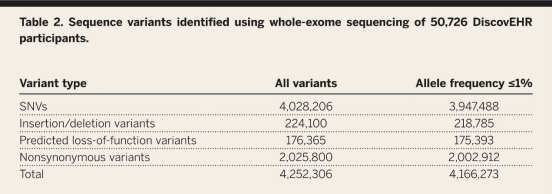
表2数据显示,在50,726名参与者中,研究识别出了超过400万个独特的单核苷酸变异和22.4万个插入/缺失变异。大多数变体(约98%)的等位基因频率均≤1%,这表明这些变异在总体人群中是相对稀有的。预测功能丧失(pLoF)变体在所有变体中的数量为176,365个,这其中约99%的变体等位基因频率≤1%,这意味着这些变体对基因功能有显著的潜在影响,并且在总体人群中主要以低频率存在。非同义变体的数量超过200万个,其中99%的变体等位基因频率≤1%。
图1 50,726个外显子序列中功能变体的频率和分布
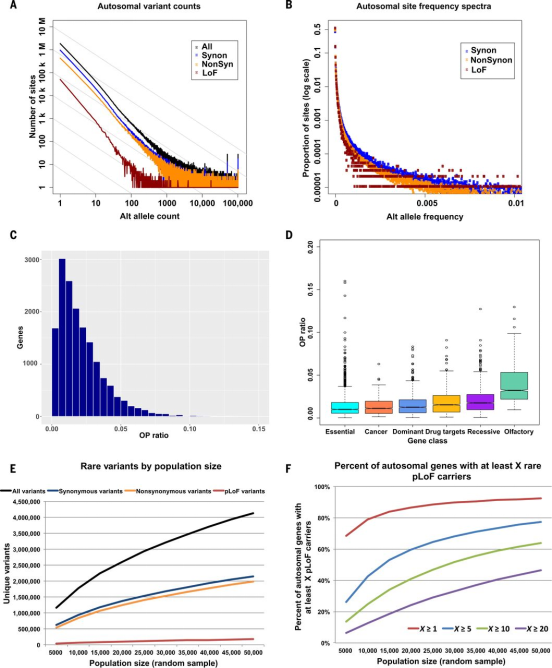
图1A显示了在不同功能类别中(同义变体Synon、非同义变体NonSyn、功能丧失变体LoF)的SNV位点数量与替代等位基因数量的关系,其中LoF变体主要集中在稀有等位基因频率(即替代等位基因数目较少)的区域。
图1B则比较了不同功能类别的SNV位点频率频谱,展示了更具功能破坏性的变体在稀有变体中的富集。
图1C展示了50,726个基因外显子序列中预测早停变体的OP比率分布情况,大多数基因的预测早停变体OP比率较低,表明这些变体在基因组中的分布受到强烈的净化选择压力。 图1D比较了不同基因类别(如必需基因、癌症基因、常显疾病基因、药物靶点基因、常隐疾病基因和嗅觉受体基因)中预测早停变体的OP比率分布,其中必需基因和癌症基因对pLoF变体的容忍度最低,常隐疾病基因、药物靶点基因和嗅觉受体基因对pLoF变体的容忍度则较高。
图1E展示了随着样本量增加,各功能类别的稀有变体(频率小于1%的替代等位基因)的累积情况,表明随着样本量的增加,研究者发现的稀有变体的数量显著增加,且pLoF变体的比例显著增加,而Synon和NonSyn变体的比例则基本不变。
图1F展示了随着随机抽样的样本量增加,至少携带1个、5个、10个和20个稀有pLoF变体的基因比例,结果发现随着样本量增加,携带pLoF变体数量越多的基因增长的比例就越大,这表明越是大规模测序越能发现罕见的pLoF变体并实现对这一类变体的深入分析。
表3 在50,726名DiscovEHR参与者中等位基因频率≤1%的预测功能丧失变体影响的基因数量
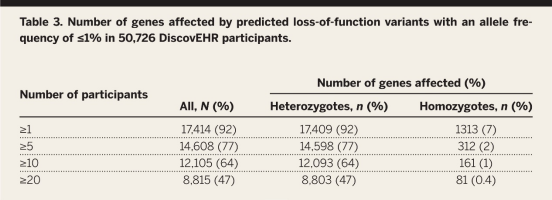
表3数据显示,在17,414个基因(约92%)中至少有一名罕见的pLoF变体携带者,其中绝大多数pLoF携带者是杂合子。这表明,相较于异质变体,纯合变体的频率和数量更低。
图2 外显子组水平和空腹血脂水平的关系

图2A展示了等位基因频率与效应大小的关系。图中信息表明在外显子组水平的关联测试中,稀有变体往往具有更大的效应大小,特别是对于甘油三酯、低密度脂蛋白胆固醇(LDL-C)、高密度脂蛋白胆固醇(HDL-C)和总胆固醇等血脂值。
图2B则具体展示了G6PC基因中多个pLoF和有害错义变体的血脂值,表明G6PC基因中的变体携带者显示出更高的甘油三酯水平。
图3 pLoF变体与脂质药物靶点基因和血脂水平的关系
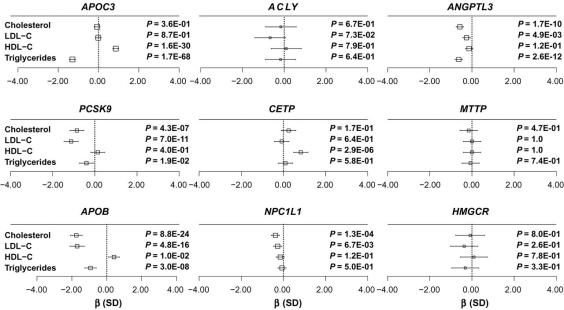
图3展示了九种(APOC3、ACLY、ANGPTL3、PCSK9、CETP、MTTP、APOB、NPC1L1和HMGCR)针对脂质药物靶点基因中的pLoF变体与不同血脂指标(总胆固醇、低密度脂蛋白胆固醇 LDL-C、高密度脂蛋白胆固醇 HDL-C、甘油三酯)的关联。六个药物靶点基因(APOC3、ANGPTL3、PCSK9、CETP、APOB和NPC1L1)中的pLoF变体与血脂水平改变显著相关,总共在36次测试中有15次名义上的显著关联(P < 0.05)。表明理论上当这些呈现显著相关性的药物靶点基因在其功能被抑制时,或许能对临床治疗产生影响。
总结
DiscovEHR项目通过对50,726名参与者的高通量外显子组测序和EHR数据的整合,得出了许多重要的科学发现和临床意义。该研究鉴定出约420万个单核苷酸变异(SNVs)和插入/缺失事件(indels),其中约17.6万个变体预计会导致基因功能丧失(LoF)。每个参与者平均携带21个罕见的、预测会丢失功能的基因变体,这表明在人类基因组数据中存在大量罕见的功能丧失变体。此外,约3.5%的参与者带有76个与临床疾病相关的基因中的致病或可能致病变体。通过对EHR数据的回顾,发现这些变体与大约65%携带者的病历记录中的单基因病相关联。
除了这些整体性数据,研究还发现CSF2RB基因中的pLoF变体与嗜碱性粒细胞和嗜酸性粒细胞计数之间存在关联,EGLN1基因和红细胞增多症之间存在家族性关联,G6PC基因中存在与血脂水平(如甘油三酯)相关的罕见变体等数个基因变体层面的信息,为进一步理解基因变体在人体中具有的重要生物学作用提供了直接证据。
DiscovEHR研究展示了将大规模基因测序与综合健康系统相结合的独特优势,验证了这类大规模基因组研究在揭示人类遗传变异的临床影响方面的潜力,并为新药开发和治疗提供了宝贵的基因靶点。
然而,研究也指出了大规模基因组测序在临床应用中面临的挑战。首先,数据处理和管理是一个巨大的技术挑战,特别是在应对海量测序数据时,需要高效的计算资源和先进的生物信息学工具。其次,变体功能预测与注释的准确性也需要进一步提升。尽管使用了先进的注释工具,预测的功能影响仍需实验验证以确保其临床相关性。
研究还强调了在大规模基因组测序中,理解和管理罕见变体的生物学和临床重要性。由于这些变体频率极低,需要更大规模的样本和详细的临床表型数据,才能充分了解其影响。EHR数据的整合提供了一个强大的工具,使得研究人员能够深入挖掘基因与表型之间的复杂关系,但也需要标准化和结构化的临床数据来支持高质量的关联研究。
最终,DiscovEHR研究的成果验证了精准医疗的巨大潜力,通过遗传筛查提前识别高风险人群,并通过个性化治疗提升临床效果。这项研究为未来的基因组医学实施提供了宝贵的经验,也为精准医疗的大规模应用奠定了科学基础。研究团队建议,未来的工作需要继续推进技术进步和跨学科合作,以克服目前的挑战,并充分发挥基因组数据在临床实践中的潜力。
参考文献
将结合双方在基于人工智能(AI)的基因组解读和基因组分析技术优势及行业专长,加速药物靶点的发现...

此次史无前例的合作将利用基因组学和人工智能技术识别药物靶点,加速开发新疗法...

结合影像、多组学技术和大数据,发现常见慢性疾病的新诊断方法和药物作用靶点 ...

这项为期多年的协议旨在通过大规模基因组学和建立卓越的临床基因组资源来加速治疗方法的开发...
旨在通过大规模基因组学研究和建立卓越的临床基因组资源来加速药物研发进程...