
2025年2月21日
在测序仪读取DNA样本之前,必须经历一个漫长的多步骤工作流程,称为文库制备:首先,科学家对DNA进行定量和归一化,确保样本量适用于所用的仪器。然后,研究人员将DNA片段化成数千个片段,每个片段长300至500个碱基对,通过变性步骤将双螺旋结构分裂成单链,并在每个片段的末端标记合成的接头分子。这些接头随后将在测序运行的簇生成步骤中使用。
所有“短读长”文库制备都涉及到这些步骤。(长读长技术也存在,但需要更广泛的设置。)一些方法使用超声波振动来剪切DNA,这可能需要五个多小时。而因美纳文库制备使用的是被合成分子(称为转座子)覆盖的微小磁珠。转座子在单一步骤中将DNA片段化并标记——“酶切法片段化”。该方法使用较少的设备,并将制备时间缩短至约两小时。之后,如果DNA的可用量非常少,则PCR扩增步骤可以使样本更容易被测序仪读取。
以上仅为主要步骤,整个工作流程还包括多个清洗和纯化、稀释以及验证步骤。只有在所有的文库制备完成后,DNA才会被移入卡盒中,并流过流动槽,簇生成步骤会进一步扩增片段,以便测序仪读取。其中的每一个步骤都自然地构成一个接触点,可能会发生潜在的错误或误操作,即使是训练有素的研究人员也会出现失误。
但因美纳的最新突破,即Constillation已定位读数技术,完全省去了这种工作流程。文库制备现在可直接在流动槽上进行,无需任何准备步骤。
因美纳科学研究副总监兼Constitutation项目负责人Louise Fraser说:“这很容易做到,您不需要专业的文库制备培训。见识过该技术的内部和外部人员都惊讶于它的简便性。”
因美纳生物信息学高级经理Mitch Bekritsky总结道:“提取DNA,上样至测序卡盒中,将卡盒放到测序仪上,再向卡盒中加入一些其它试剂,这样就完成了。您就大功告成了。提取完成后,全程大约需要10至15分钟。这个过程非常复杂,繁琐,但它使其更加简单。”
此外,由于DNA在流动槽上固定就位后才会发生片段化,新的生物信息学算法可以利用片段的物理位置来推断它们在原始序列中的位置,获得之前认为只有长读长技术才能实现的远程见解。
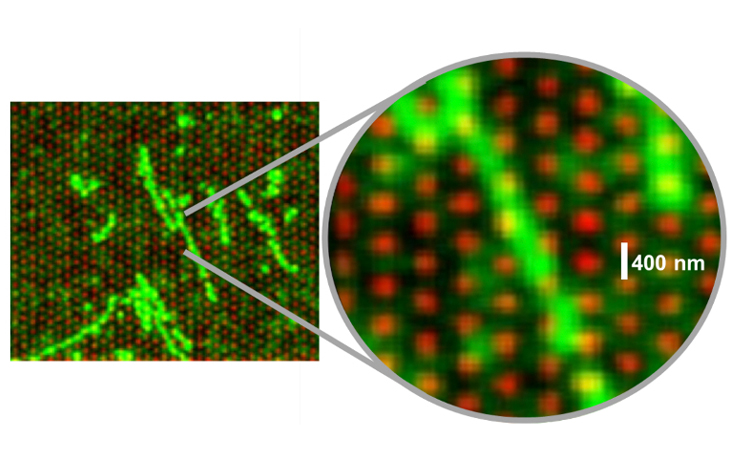
该荧光图像显示了完整的DNA链如何穿过流动槽表面的多个纳米井而沉降。| 图片来自因美纳
连接点
多年来,因美纳的科学家们一直在开发Constitution已定位读数技术,但直到最近才实现应用。
现用的因美纳流动槽是由数十亿个微小的纳米井组成的。这些纳米井确保簇以特定且规律的间隔生成,以及测序仪确切地知道读取位置。早期的流动槽设计是未阵列化的,因此簇会在完全随机的位置生成,这增加了读取它们所需的时间和精力。
Louise Fraser在因美纳工作的18年里大部分时间都在努力让文库制备变得更快、更容易。实际上,她的团队很久之前就成功地利用附着在流动槽表面的转座子完成了流动槽内片段化,但这只适用于当时所用的未阵列化、随机排列的流动槽。当新的流动槽引入纳米井后(在随后的模型中,纳米井变得更小、更紧密),面临的挑战是如何精确设计转座子使其以恰好的浓度最终进入每个纳米井中。
因美纳研发团队在2023年再次接手了这个Constellation项目。他们创建了一种包含三种试剂的定制方案,用户可将其添加到卡盒中使其运作:首先,使用缓冲液稀释DNA样本,并制备用于片段化。其次,是一种有助于转座子粘附到纳米井上的混合物。第三种试剂含有转座子本身。这三种试剂的组合使用户能够将完整的双链DNA直接流过流动槽表面。转座子沉淀在纳米井中,锁定到DNA上,对其进行标记,然后片段生成簇,测序工作流程如前所述。
然后,在开发过程的某一刻,研发团队看到DNA以蛇形模式在流动槽表面来回沉降,于是他们灵光一闪:由于完整的DNA链在片段化之前跨越多个纳米井,因此有理由认为来自邻近簇的读数可以追溯到基因序列中的相邻位置。
这是一个意想不到但具有革命性的流动槽内片段化特性。仅使用一组定制试剂,无需对测序仪器进行任何其他调整,研究人员就可利用簇邻近信息检测短读长技术无法检出的较大基因组变异。
Mitch Bekritsky所在团队开发了DRAGEN二级分析软件的算法,以便利用这些信息。他们建立了概率模型,用于确定从物理上的近端纳米井中读取的结果来源于近端基因组区域的可能性。该算法连接纳米井之间的“点”,有效地重新描绘原始DNA链的形状。这些连接所揭示的模式是这项新技术名称的灵感来源:Constitution。
更完整的基因组
短读长测序面临的最大挑战是基因组的高度重复或同源区域。当分析软件试图找出短片段如何拟合在一起时,这些重复区域会模糊地拟合到多个可能的位置,导致匹配置信度低。而一些感兴趣的突变,如大型的倒位或结构变异,只能通过一次观察很长的片段来检测。
直到现在,人们还认为只有长读长技术才能准确定位这些变异,但得益于Constitution赋予的邻近信息的额外维度,DRAGEN软件现在可以定位以前无法定位的读数。该技术可使假阳性和假阴性单核苷酸多态性(SNP)的检出率降低40%。
Constellation也被证明非常适用于定相,或将SNP分配至一种单倍型或另一种单倍型。每个人都有两种单倍型——一组基因遗传自母亲,另一组遗传自父亲。对于许多遗传病,如果个体某个基因的两个拷贝都是非功能性的,就会患上这种疾病,但如果有一个拷贝是功能性的,则会成为疾病携带者。传统的短读长技术很难定相,也很难确定无功能基因是仅来自个体的单倍型之一,还是来自两者。Constellation也能弥合这一差距,将超长序列定相至数百万个碱基对。
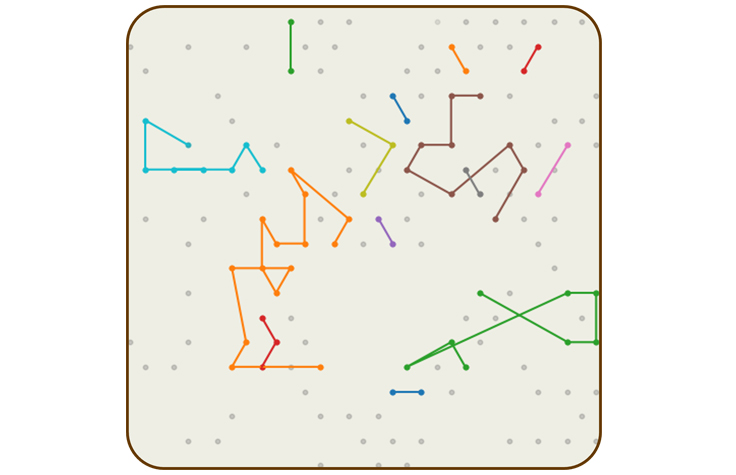
Constellation技术使用概率模型来推断来自相邻流动槽的读数如何在原始序列中连接。| 图示来自因美纳
真实的结果与影响
在2024年11月的美国人类遗传学学会年会上,因美纳的首席技术官Steven Barnard和Broad Clinical Labs的首席科学官Niall Lennon谈到了他们的科学家在那个夏天合作进行了Constillation技术的测试运行。Lennon称赞了它的易用性,说:“如果您已经在基因组学技术领域工作了一段时间,就会记得许多团队都做出了这样的承诺——这是19年来我第一次看到有人真正提出了一种无需文库制备的方法。无需接头连接。只需将DNA置于测序仪上,它就能工作。”
Bekritsky分享说,关于Constellation开发,他最欣赏的一点是,它需要每个部门的团队共同努力。“对于一家只做文库制备、只做测序,或只做信息学分析的公司来说,那就不可能了。而在因美纳,我们召集了所有相关人员共同开发。”
这项技术可能产生的潜在现实影响让他深受启发——他们测试的第一批样本中有一些来自儿科血友病患者,这项技术检测到了其中存在的每一个基因倒位。Bekritsky记得他看到这些结果后十分惊讶。“这就不是抽象的概念了。”他说,“我们实际上可以解决这些倒位问题,并且确切地说,这就是这些孩子患有血友病的原因。感谢这个能够实现创新的项目。”
首款采用Constellation技术的商业化产品预计将于2026年推出,与NovaSeq™ X和NovaSeq™ X Plus测序仪兼容。 ◆
如需了解更多有关Constillation已定位读数技术的信息,请阅读 Illumina Genomics Research Hub上的这篇文章。
2025年2月24日:这篇文章的早期版本错误地将Constitutium技术称为使用“转座子磁珠”。虽然传统的文库制备确实使用了磁珠,但使用Constellation技术的试剂却未使用。此错误已更正。


