Summary
The LPA KIV-2 domain remains a challenging genomic region to characterize, despite modern advances in sequencing technology—but considering this region’s impact on human health, it is essential to improve understanding of its variability. Here we describe a new DRAGEN targeted copy number calling strategy for the Kringle-IV type 2 (KIV-2) domain of LPA. The DRAGEN KIV-2 caller makes a major improvement to the state of the art in LPA characterization and reveals important insights about the variability of the region. While challenges remain in fully comprehending KIV-2 variation and its impact, this software will provide a valuable tool for enhanced LPA and CVD research, furthering the scientific mission of understanding human genetic disease and advancing the goal of achieving a fully featured genome. The DRAGEN KIV-2 caller will be featured in a future release of the Illumina DRAGEN genome analysis pipeline.
For further reading that goes into greater detail, please see this recently published article at BioRxiv.
Introduction
Decreased KIV-2 copy number → increased Lp(a) concentration → increased CVD risk
Cardiovascular disease (CVD) is the leading cause of mortality globally and has a complex array of known and postulated causes.1 One important risk factor for CVD is abnormally high blood plasma concentration of lipoprotein(a), also called Lp(a), a low-density lipoprotein.2 Elevated Lp(a) concentration has been strongly associated with increased CVD risk.3 Lp(a) levels are highly heritable and elevated Lp(a) levels are also extremely common, occurring in one out of five individuals.4 Approximately 69% of Lp(a) concentration variability is driven by the copy number of the KIV-2 domain in the protein.5 The KIV-2 domain is encoded in the genome by a 30 kilobase (kb) region of the LPA gene, including two introns and two exons, which occurs as a six-copy repeat array in the GRCh37/38 reference genomes, totaling 180 kb of reference sequence (Figure 1A). This repeat array is considered a variable number tandem repeat (VNTR) and it exhibits marked copy number variability throughout the population.6,7
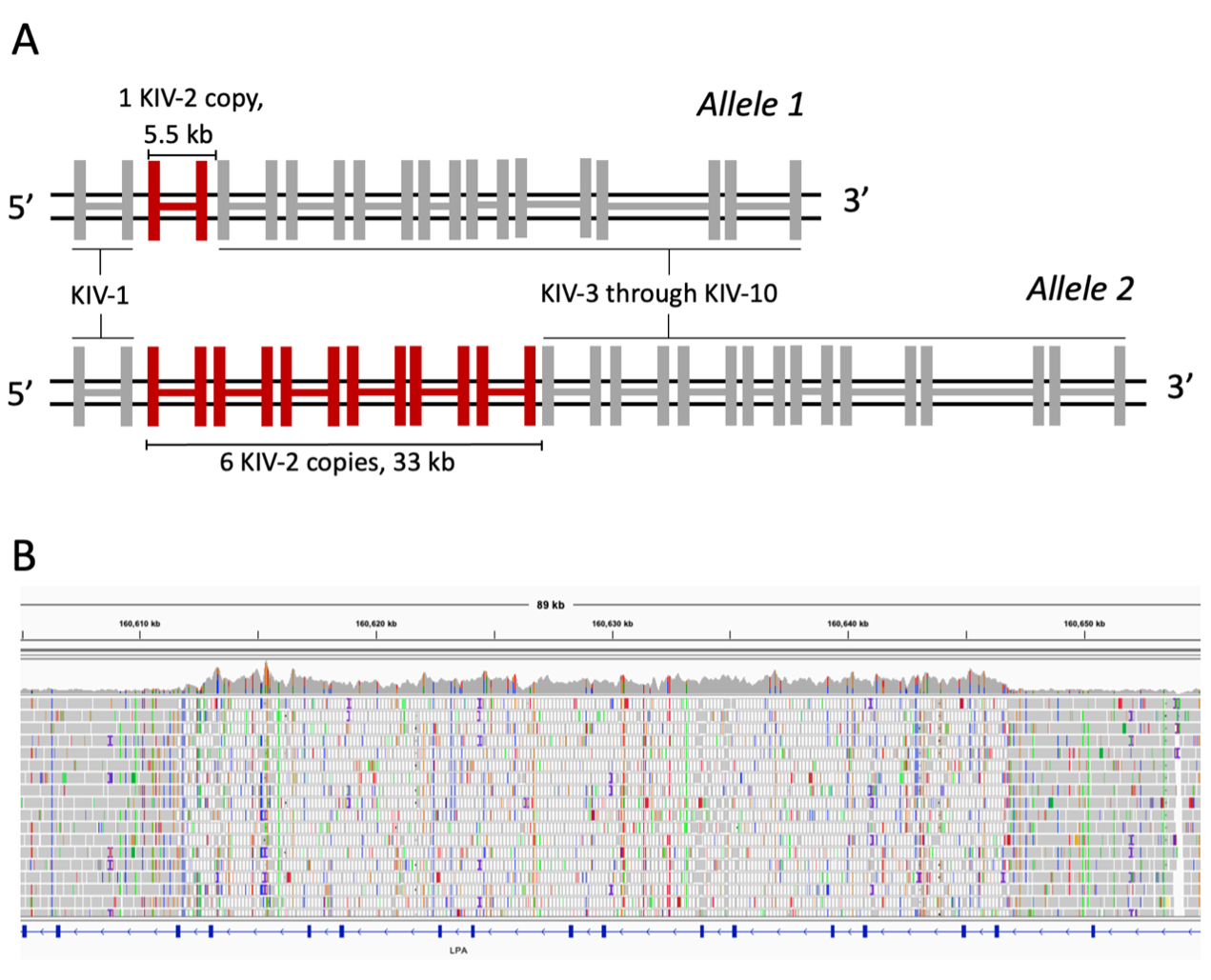
Figure 1A. A diagram of the KIV-2 region of LPA, showing two alleles. Kringle IV domains are represented as connected pairs of vertical bars as LPA exons. In allele 1 a single copy of KIV-2 is shown in red, and the approximate length of the repeat unit is shown. In allele 2, six copies of KIV-2 are shown, corresponding to the number of copies in the GRCh37/8 reference genomes, with the approximate length of the repeat array shown.
Figure 1B. An Integrative Genomics Viewer screenshot of the KIV-2 region in Genome in a Bottle sample HG002. An increase in coverage occurs in the center of the view, corresponding with the KIV-2 region and indicating an alteration of copy number relative to the reference genome. The mapping quality of reads also drops to zero for much of the region, indicated by white reads. The low mapping quality shows that reads are mapped with equivalent confidence to multiple locations, illustrating the repetitive nature of this region.
Increased copy number of the KIV-2 repeat array is associated with decreased Lp(a) concentration, likely due to decreased secretion of long Lp(a) isoforms by the endoplasmic reticulum.8 Thus, low KIV-2 repeat copy number is associated with elevated Lp(a) levels and increased risk of CVD.
The impact of KIV-2 copy number makes it a very important genomic feature to measure, but the large size and population variation of the region confound accurate quantification. Reads generated from whole-genome sequencing (WGS) fail to align accurately to a single location in the reference genome (Figure 1B), and the potential for extremely high copy number is challenging for standard copy number identification strategies.
Method
Here we present a new DRAGEN targeted WGS-based copy number calling strategy for KIV-2. This approach uses the counts of reads mapped to any copy of the KIV-2 region, performs GC-correction by internal normalization to a panel of other genomic regions, and scales the resulting value to generate a highly accurate total copy number measurement.
Phased allele length calculation is performed by leveraging a pair of intronic marker sites where analysis has revealed that the reference and alternate nucleotide bases are identical in every copy of the KIV-2 domain within the same LPA allele. That is, if an inherited allele of LPA contains the reference bases at the marker sites in any copy of the KIV-2 repeat, every other copy of KIV-2 within that inherited LPA allele will also contain the reference bases for the marker sites. Proportional measurement of allelic, or phased, copy number for each of the inherited LPA alleles is therefore possible when the markers are inherited heterozygously, by leveraging the proportion of reads containing the reference or alternate alleles for the marker sites.
Results
We tested the DRAGEN KIV-2 caller by calling copy number in family trios (one offspring and two parents sequenced) and duos from the Extended 1000 Genomes (1KG) cohort9 by assessing for Mendelian consistency of inherited alleles. In 120 trios, where we determined allelic copy number for all three samples, we matched DRAGEN KIV-2 allelic copy number calls from both offspring alleles to the closest-matching parental copy. The Pearson’s correlation between the offspring and matched parental KIV-2 allele lengths showed a very strong correlation coefficient of 0.998 (P ≈ 3.58e-98). We included 1KG population data in the comparisons between offspring and parent allele lengths (Figure 2A-B), showing that all major populations have similar offspring-to-parent allele length consistencies.
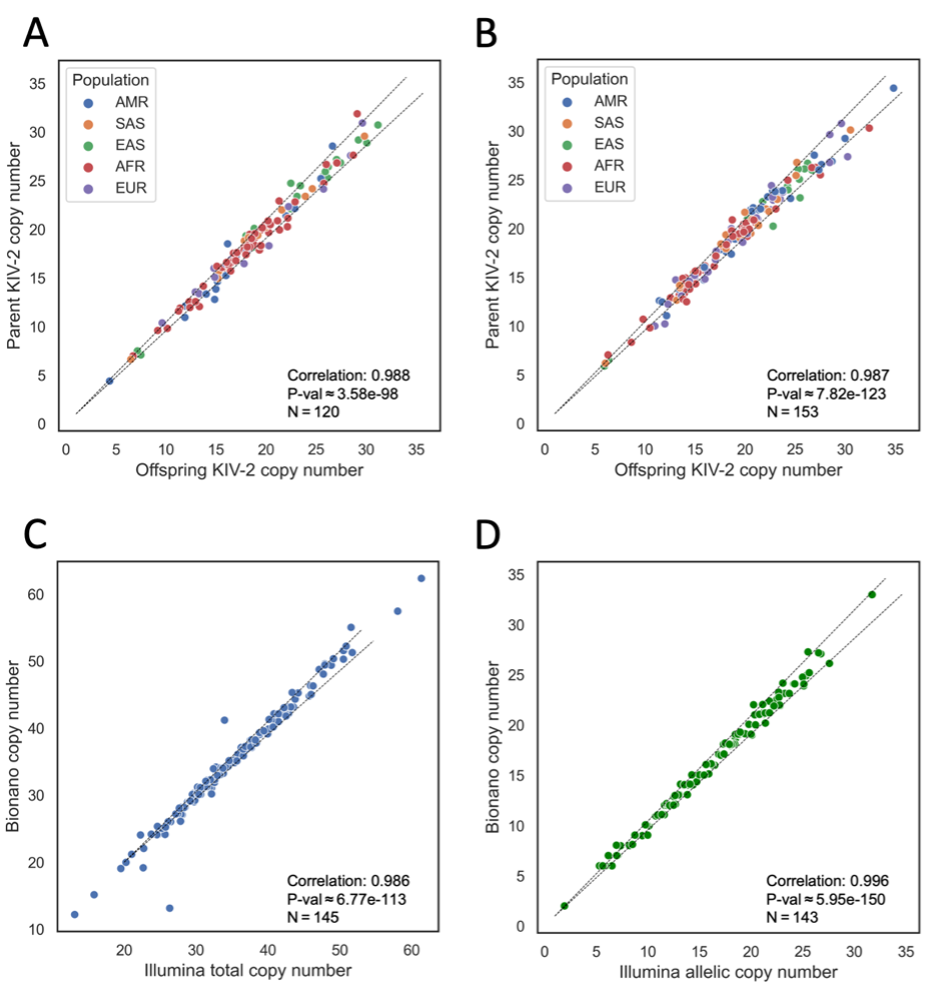
Figure 2A. Trio-based comparison of both phased KIV-2 copy numbers in 120 offspring to each of the most closely matching parental phased copy numbers, one from each parent, from families with both parents sequenced and one offspring sequenced. Pearson’s correlation coefficient and P-value shown at bottom right.
Figure 2B. Duo-based comparison of one phased KIV-2 copy number in 153 offspring to the single most closely matching parental phased copy number, from families with one parent sequenced and one offspring sequenced. Pearson’s correlation coefficient and P-value shown at bottom right.
Figure 2C. Comparison of total copy number calls for 145 samples against calls from Bionano optical mapping. Pearson’s correlation coefficient and P-value shown at top left.
Figure 2D. Comparison of allelic copy number calls for 145 alleles against calls from Bionano optical mapping. Pearson’s correlation coefficient and P-value shown at top left.
AMR = Admixed American, SAS = South Asian, EAS = East Asian, AFR = African, EUR = European. P-values obtained by Kolmogorov-Smirnov test.
We compared the results of total and allelic copy number calling against KIV-2 copy number calls generated by Bionano optical mapping, an orthogonal technology (Figure 2C-D). DRAGEN KIV-2 calls were compared against 145 orthogonal total copy number calls and 143 allelic orthogonal copy number calls. These correlated closely as well (0.986 with P ≈ 6.77e-113 and 0.996 with P ≈ 5.95e-150, respectively). These comparisons show that the accuracy of DRAGEN KIV-2 calls is consistent between similar alleles as transmitted through inheritance, and that the DRAGEN KIV-2 copy number calls are generally similar to calls from orthogonal technologies, highlighting the consistency and accuracy of this WGS-based approach.
Population variation of KIV-2 alleles between major populations is a factor of great importance for determining population-specific CVD risk factors. We performed preliminary analysis of this variation by calling copy number for all samples in the 1KG cohort, then comparing the distribution of calls for each population group in 1KG to the distribution of calls in all other populations combined. The AMR and SAS groups were not significantly different from all others (P > 0.05), but the other three population groups (EAS, AFR, and EUR) were significantly different from the consensus (P < 0.05, Figure 3A). As lengths of the two alleles present in a sample may be quite different, we repeated this comparison for the shorter (Figure 3B) and longer (Figure 3C) alleles present in each sample.
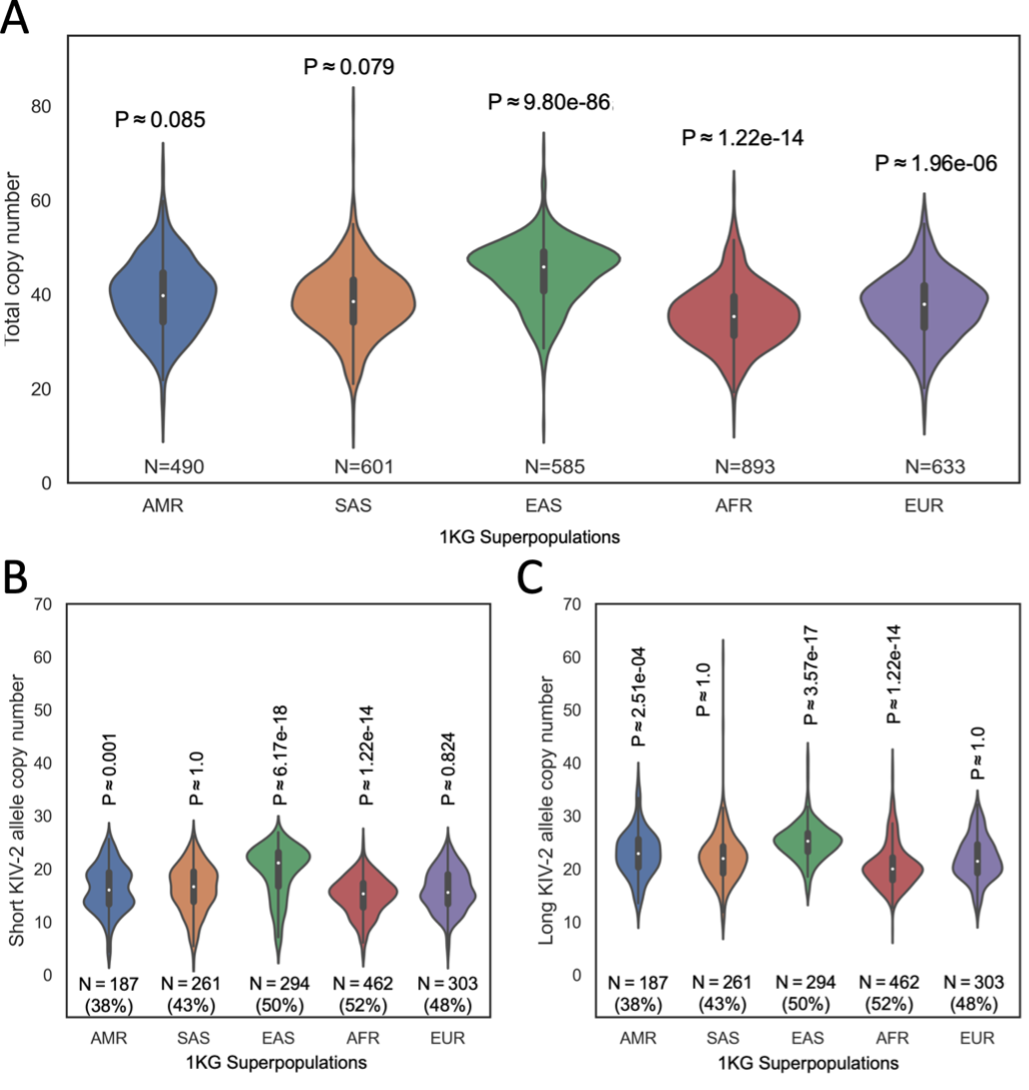
Figure 3A. The distribution of total KIV-2 copy numbers in each population group, with the count of samples in that group.
Figure 3B. The distribution of phased KIV-2 copy numbers in each population group, including only the shorter of the two alleles present in each sample. The count of samples in that group and the percentage of samples with allelic copy number calls is shown beneath each distribution.
Figure 3C. The distribution of phased KIV-2 copy numbers in each population group, including only the longer of the two alleles present. The count of samples in that group and the percentage of samples with allelic copy number calls is shown beneath each distribution.
AMR = Admixed American, SAS = South Asian, EAS = East Asian, AFR = African, EUR = European. P-values obtained by Kolmogorov-Smirnov test and adjusted for n = 5 tests by the Bonferroni method.
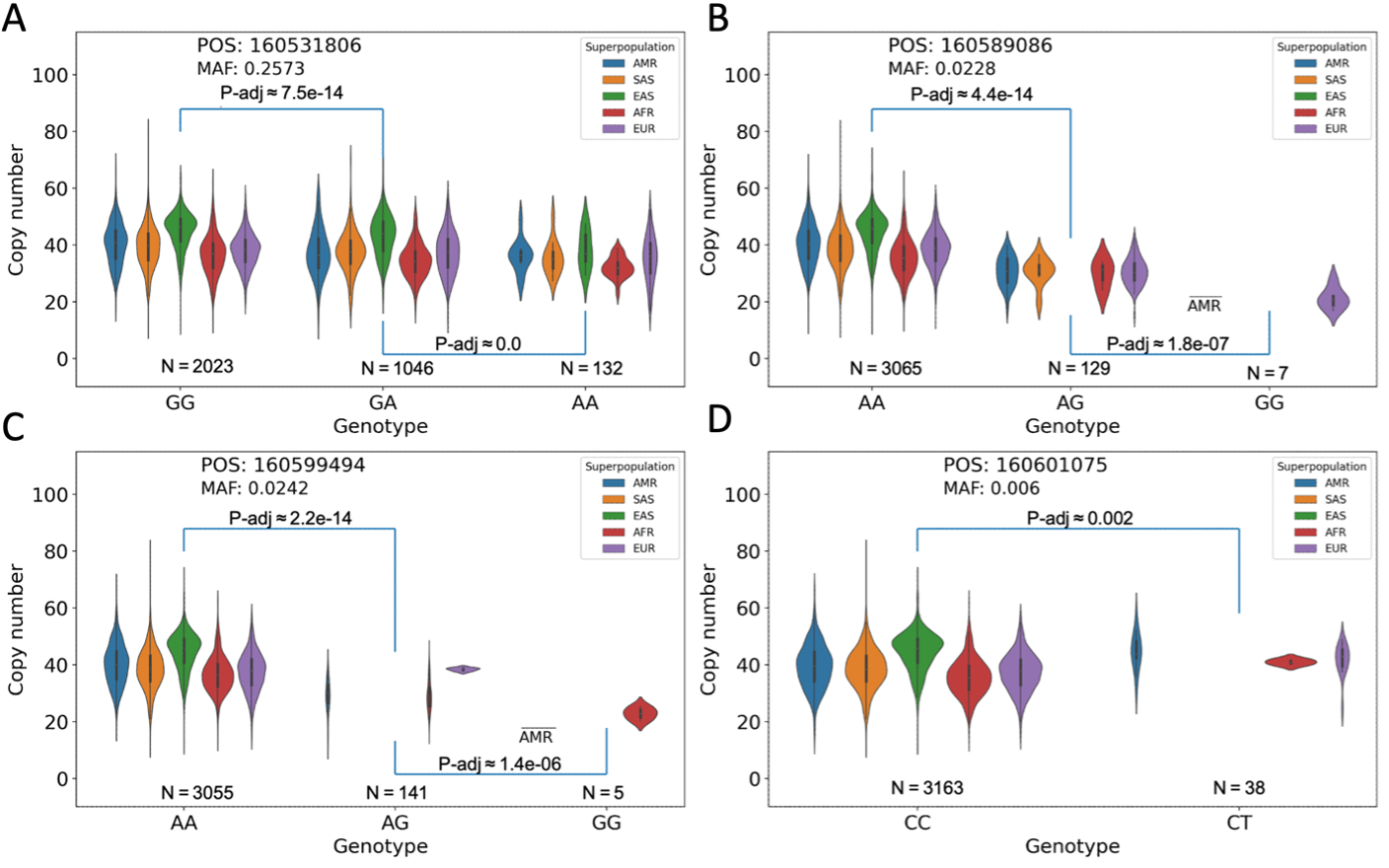
Each panel contains a population-segmented distribution plot of observed total KIV-2 copy numbers, separated into categories for homozygous presence of the most common (major) allele, heterozygous presence of the major allele and a less common (minor) allele, or homozygous presence of the minor allele. P-values are derived by Kolmogorov-Smirnov tests of the similarity between each annotated pair of distributions.
Figure 4A. The minor allele is present in all population groups and the difference between the allele-stratified distributions is significant. However, a large range of alleles is present in each distribution.
Figure 4B. The minor allele is not present in the EAS population group and is rare in all population groups, occurring homozygously in only seven samples. The range of copy numbers for those seven samples is considerably restricted relative to the major allele case.
Figure 4C. The minor allele is not present in the SAS or EAS population group and occurs homozygously in only one sample from the AMR group, plus four from the AFR group.
Figure 4D. Similar to both B and C, this minor allele marker is present in a restricted subset of populations (AMR, AFR, and EUR), although in this case it does not occur homozygously in any sample. A wide range of copy numbers is observed in the heterozygous cases.
Acknowledgments
We thank our co-authors Sairam Behera, Fritz Sedlazeck, Ginger Metcalf, Luis Paulin, Vipin Menon, and Christie Ballantyne at the Baylor College of Medicine; Xiao Chen and Michael Eberle at Pacific Biosciences; Bing Yu, Ngoc Nguyen, and Eric Boerwinkle at the University of Texas Health Science Center at Houston, Texas; and Carlos Rodriguez and Robert Kaplan at the Albert Einstein College of Medicine.
References
- Cardiovascular diseases (CVDs). https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
- McCormick, S. P. A. Lipoprotein(a): Biology and Clinical Importance. Clin. Biochem. Rev. 25, 69 (2004).
- Emdin, C. A. et al. Phenotypic Characterization of Genetically Lowered Human Lipoprotein(a) Levels. J. Am. Coll. Cardiol. 68, 2761–2772 (2016).
- Trinder, M., Uddin, M. M., Finneran, P., Aragam, K. G. & Natarajan, P. Clinical Utility of Lipoprotein(a) and LPA Genetic Risk Score in Risk Prediction of Incident Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 6, 287–295 (2021).
- Boerwinkle, E. et al. Apolipoprotein(a) gene accounts for greater than 90% of the variation in plasma lipoprotein(a) concentrations. J. Clin. Invest. 90, 52–60 (1992).
- Mukamel, R. E. et al. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science (80-. ). 373, 1499–1505 (2021).
- Afshar, M. & Thanassoulis, G. Lipoprotein(a): New insights from modern genomics. Curr. Opin. Lipidol. 28, 170–176 (2017).
- Schmidt, K., Noureen, A., Kronenberg, F. & Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 57, 1339–1359 (2016).
- Byrska-Bishop, M. et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 2021.02.06.430068 (2021) doi:10.1101/2021.02.06.430068.