关于DRAGEN STR的关键信息
- DRAGEN DNA流程包括一个短串联重复检出程序
- 与无需PCR的全基因组测序结合使用时,该检出程序对FMR1、ATXN1、HTT和C29orf72等基因中与医学相关的重复扩增的灵敏度 > 98%
- 虽然假阳性很少(<1%),但建议进行确证研究,以确认并更准确地确定推定的阳性结果
什么是STR以及为什么它们很重要?
短串联重复序列(STR)是指基因组中简单的DNA序列连续重复的区域(表1)。人类基因组中有许多STR区域,其中大多数区域的功能尚不清楚。
| 人员A:(CAT)×3:...CATCATCAT... |
| 人员B:(CAT)×1:...CAT... |
| 人员C:(CAT)×9:...CATCATCATCATCATCATCATCATCAT... |
表1.STR的假设示例
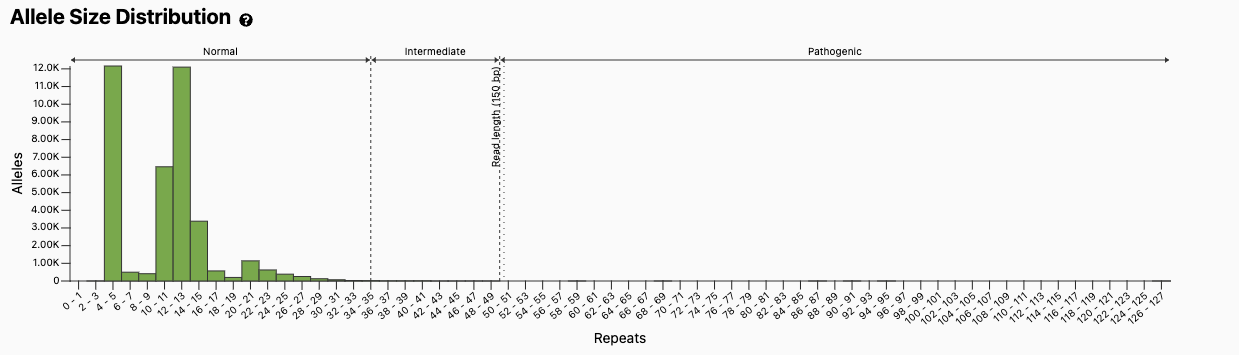
图片摘自gnomAD v3数据库, PMID 324616541。
首次发现STR变异与疾病之间存在关联的是亨廷顿病。如需了解关于Marcy MacDonald博士及其团队的突破性发现的更多信息,请查看Nature Education上的文章。亨廷顿病患者的HTT基因(以该疾病命名)中含有超过40个连续的C-A-G三核苷酸组(“三核苷酸重复”)。该序列位于基因的编码序列内,被翻译成重复的谷氨酰胺。由此产生的蛋白质中连续谷氨酰胺数量的增加会导致神经元聚集,最终导致亨廷顿病的临床体征和症状,包括共济失调和神经功能衰退。
亨廷顿病是一种常染色体显性遗传病,在亨廷顿病家族中,患者的子女往往会更早出现症状,神经退行性疾病的进程也更快。STR分析表明,在受到严重影响的后代中,扩增的重复序列进一步扩增,导致了这一被称为“早现”的悲剧现象。
亨廷顿病最狡猾的一点是,患者通常在生育后才会出现症状,而他们的孩子有50%的风险会遗传到进一步扩增的STR。利用现有的工具,从技术上来说,可以更早地对每个人进行这一疾病筛查。遗憾的是,目前尚无有效的治疗方法,因此不宜向普通人群推荐此类检测。目前正在努力研究亨廷顿病的靶向基因疗法。如果能够开发出治疗亨廷顿病的基因疗法,那么对每个人进行检测就会变得合理可行。
发现HTT重复扩增也使其他团队开始在其他病症中寻找这种变异。目前至少有56种不同基因的STR与人类疾病相关(图2),包括脆性X综合征(FMR1基因),它是一种最常见的遗传性智力障碍,也是美国医学遗传学和基因组学学会建议进行携带者筛查的最重要的疾病之一。
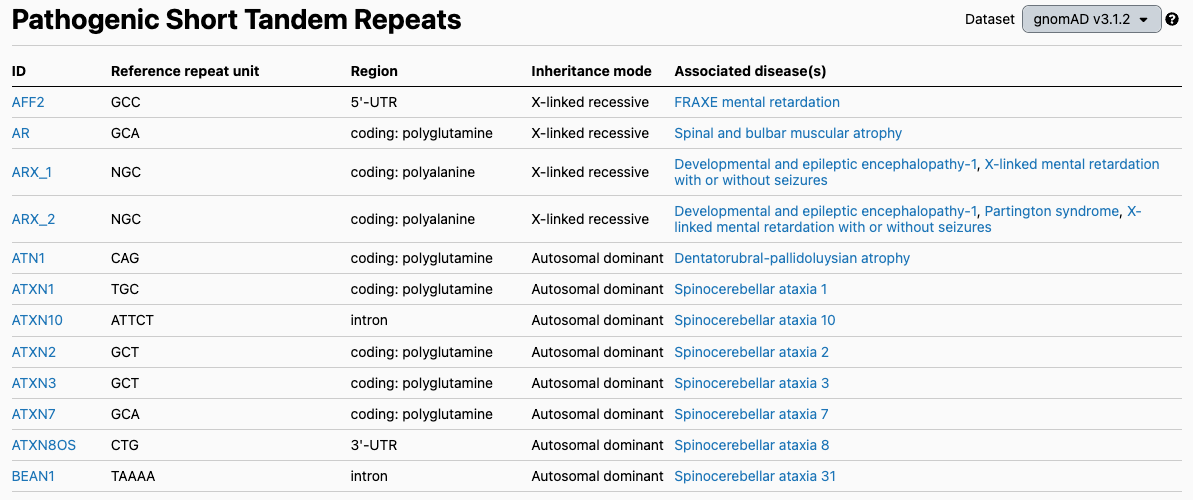
图片摘自gnomAD v3数据库,PMID 324616541。
STR通常如何分析?
评估STR的第一种有效方法是Southern blot。虽然这种方法灵敏度高,但在实验室中操作起来很麻烦,而且很难准确评估重复次数(图3)。
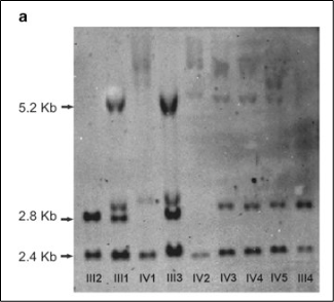
截图摘自PMID 211073402。原标题:“FMR1(脆性X智力障碍1)基因的Southern blot分析。显示了正常未甲基化(2.8 kb)、正常甲基化(5.2 kb)和对照条带(2.4 kb)的大小。”
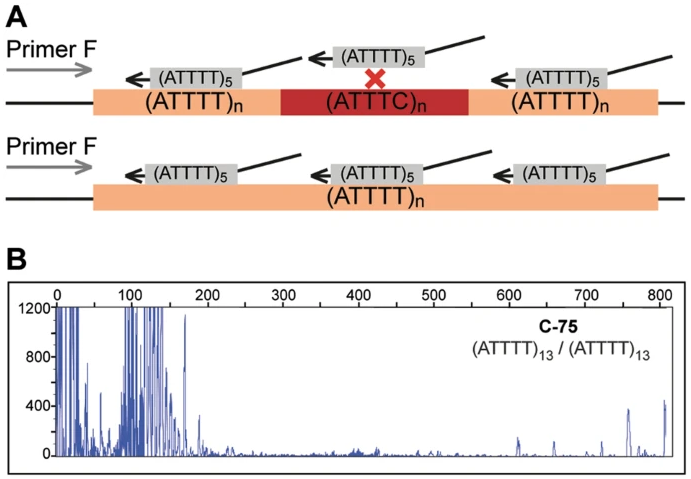
截图摘自PMID 298919313。原标题:“ATTTT RP-PCR检测DAB1中的大片段五核苷酸等位基因。a ATTTT RP-PCR引物与重复的ATTTT区域结合,导致正常和突变等位基因中的DNA扩增的示意图。b 电泳图,显示了表1中对照组个体(C-75、C-88、C-91、C-95 和 C-44)以及SCA37患者A-1和A-9的荧光ATTTT RP-PCR分析结果”
可以使用panel或外显子组数据分析STR吗?
非常遗憾,对于某些重复模式,NGS panel和外显子组测序所必需的文库制备和靶向扩增或杂交过程会将重复性DNA从检测中去除。再多的生物信息学方法也无法挽救试管中没有的信号。尽管如此,最近的研究表明,基于富集的NGS可能适用于某些基因位点并能提高基于外显子组的检测的诊断率。探针捕获和PCR扩增的效果在所有STR模式中并不相同。虽然这种行为尚未在所有已知的STR基因位点中得到全面表征,但普遍认为100%GC基序非常难扩增。
在什么情况下会用到DRAGEN STR?
与panel或外显子组测序相比,无需PCR的全基因组测序(pfWGS)保留了重复的基因组DNA进行测序。当最相关的扩展等位基因通常超过短读长因美纳测序数据的读长时,对研究研究人员而言,面临的挑战就变成了对重复长度进行基因分型。为了解决这个问题,因美纳校友Egor Dolzhenko博士、Michael Eberle博士及其同事开发了ExpansionHunter。首先,他们为重要的STR区域创建了一组自定义参考,以便与受试者数据进行比较。该算法可识别pfWGS数据中包含丰富信息的序列read,例如侧翼区域的read和包含重复序列的read以及它们的配对read。将专门准备的参考与这些read相结合,该算法可以轻松识别未扩增的等位基因并标记可能扩增的病例。
因美纳的DRAGEN二级分析包括使用ExpansionHunter进行STR基因分型,适用于任何具有pfWGS数据的样本。这可以在本地硬件或云端运行。如果您有兴趣使用自己的数据实施DRAGEN工作流程,请联系我们!
对于那些对数学和生物信息学感兴趣的人来说,原始文献是一个很好的资源。之前的出版物对此进行了更详细的介绍。
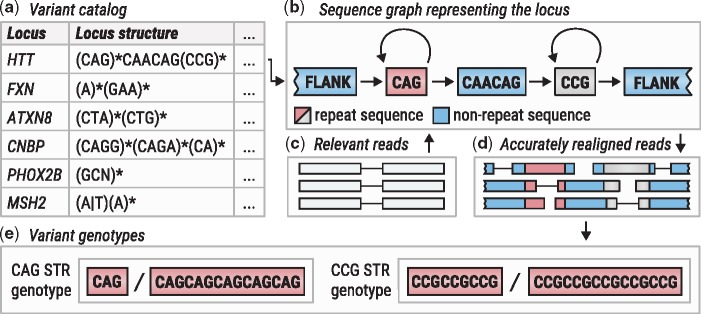
图片摘自PMID 311342794。原标题:“ExpansionHunter概览。(a) 从变异目录文件中读取基因位点定义。(b) 根据变异目录中的说明构建序列图。(c) 从输入的二进制比对/定位文件中提取相关read。(d) read与图谱比对。(e) 将比对结果拼凑在一起,对每个变异进行基因分型”
DRAGEN STR的性能如何?
当面对多种STR扩增条件的一系列阳性和阴性数据集时,ExpansionHunter表现出了优异的性能(图6)。除一个阳性对照外,所有阳性对照检测结果均为阳性,显示出非常高的准确性。尽管正常对照的检测结果呈阳性,但阴性预测值(检测结果呈阴性的真阴性比例)也非常高。这些研究结果有力地支持了“筛查和确认”方法,即对所有参与者进行pfWGS检测,然后对那些在某些基因上超过实验室验证的临界值的参与者,使用rpPCR来确认潜在的基因扩增。
图6中的研究中唯一的假阴性值得进一步讨论。此示例存在FMR1预突变,但使用预定义的临界值时被视为是正常的。这表明临床实验室可能需要考虑使用稍低的临界值并适度接受更高一些的rpPCR确认率,以确保对这类扩增实现更高的灵敏度。这是平衡灵敏度和特异性的一个典型例子。
作为开发用于全基因组测序的体外诊断设备的一部分,我们以类似的方式评估了具有临床关联性的11个位点的157个扩增等位基因和每个STR基因位点的约700个已知阴性等位基因。在这项研究中,灵敏度为98%,每个样本的假阳性率< 1%。当使用更宽松的ATXN1和ATXN2临界值时,灵敏度提高到100%。

截图摘自PMID 311342794。原标题:“对含有已知重复扩增的Coriell样本进行分析。蓝色、橙色和红色矩形分别定义了相应重复的正常、预突变和完全扩增的预期大小范围。每个点对应最长等位基因的大小,其颜色根据实验确定的状态设置。GangSTR仅在提供了预定义脱靶基因位点的STR上运行。除了FMR1之外,其他所有基因的GangSTR值都是使用“全基因组”模式计算的,而FMR1是使用“靶向”模式进行分析的,该模式对于此重复具有更好的性能。重复大小的上限为600 bp。”
英国十万人基因组计划团队重复了这项研究,并评估了该人群规模数据集中的STR(PMID:35182509)5。在本研究中,通过WGS和Expansion Hunter,从404名神经系统表型与扩增性疾病一致的个体中鉴定出了潜在的STR扩增(图7)。通过PCR对潜在的阳性结果进行了确认。他们的结果令人印象深刻:
“全基因组测序对221个扩增等位基因中的215个和1321个非扩增等位基因中的1316个基因实现了正确分类,与PCR检测结果相比,在13个疾病相关基因位点中表现出97.3%的灵敏度(95% CI 94.2-99.0)和99.6%的特异性(99.1-99.9)。”

截图摘自PMID 351825095。原标题:“使用全基因组测序检测重复扩增的性能——泳道图显示了在检测出的793个扩增中,ExpansionHunter预测的重复扩增的大小。每个基因组由两个点表示,每个点分别对应于每个基因位点的一个等位基因,但男性X染色体上的基因(即FMR1和AR)除外,这些基因仅显示一个点。点表示ExpansionHunter目视检查后估计的重复长度,颜色表示PCR评估的重复大小(蓝色代表未扩增;红色代表扩增)。这些区域以阴影表示每个基因的未扩增(蓝色)、预突变(粉色)和扩增(红色)范围,如附录(第28页)中所示。粉色或红色阴影区域中的蓝点表示假阳性,蓝色阴影区域中的红点表示假阴性。附录中提供了个体检测结果(第27页)。”
是否有可能发现新的STR基因位点?
为了极大地提高覆盖的疾病范围并支持研究人员解决未确诊的疾病,ExpansionHunter团队设计了一种新方法来识别可能的STR扩增,通过搜索基因组来查找重复read的堆积,然后比较受影响个体和一组对照样本之间这些堆积的覆盖率和位置。使用这种名为ExpansionHunter Denovo的新工具,“重新发现”了弗里德赖希共济失调和脆性X等经典STR病症(图8)。总体而言,使用这种方法,44个已知扩增中的41个被确认为阳性。计划在即将发布的DRAGEN版本中推出此软件包的重新设计版本;如有兴趣了解更多信息,请联系我们。
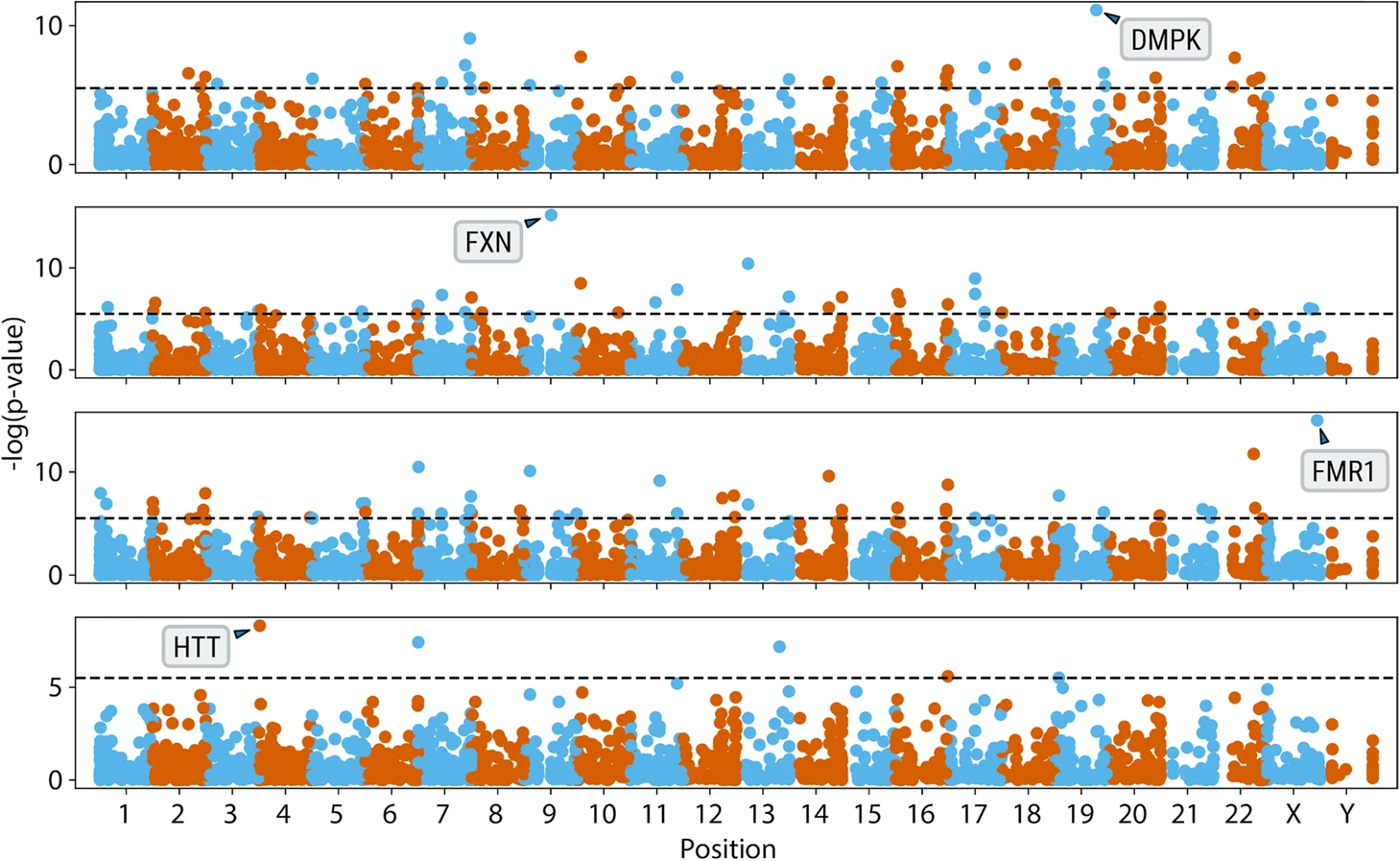
图片摘自PMID 323453455。原标题:“对锚定IRR进行全基因组分析,将DMPK、FXN、FMR1和HTT基因(从上到下)中具有已知致病性扩增的病例与150例对照进行比较”
如何进行STR检测?
运行ExpansionHunter的两种主要方式是作为DRAGEN工作流程的一部分或作为独立软件运行。DRAGEN DNAseq流程3.7.5或更高版本包含执行ExpansionHunter分析的选项(请参阅在线帮助了解详情)。DRAGEN可以使用物理硬件(“本地”)运行,也可以作为基于云的工作流程的一部分在多个平台上运行。该软件也可在GitHub上作为独立软件包提供(表2)。
| 平台 | 类型 | 说明 | 链接 | ||||||
| DRAGEN二级分析 | 本地服务器* | 定制设计的计算机硬件针对二级基因组分析(比对和变异检出)的准确性和速度进行了优化。 | https://www.illumina.com/products/by-type/informatics-products/dragen-bio-it-platform.html | ||||||
| Emedgene | 云端 | 基于云的基因组分析平台,包括panel、外显子组和全基因组。这包括DRAGEN二级分析、注释、筛选和三级分析工作流程、知识数据库、强大的报告工具以及基于人工智能的变异优先级排序。 | |||||||
| BaseSpace Sequencing Hub | 云端 | 专为管理因美纳测序运行和分析而设计的基于云的生物信息学平台。 | |||||||
| Illumina Connected Analytics | 云端 | 专为跨项目和跨类型的数据管理和分析而设计的基于云的生物信息学平台。 | https://www.illumina.com/products/by-type/informatics-products/connected-analytics.html |
||||||
| TruSight软件套装 | 云端 | 用于外显子组和全基因组端到端基因组分析的基于云的平台。 | https://www.illumina.com/products/by-type/informatics-products/trusight-software-suite.html | ||||||
| Linux | 软件 | 原始的ExpansionHunter软件包可供研究使用,并可在您自己的服务器上执行。 |
*“本地”是指安装在服务器机房/机柜中的“本地”计算机硬件。
表2.
参考文献
1. Karczewski, K.J., Francioli, L.C., Tiao, G. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). https://doi.org/10.1038/s41586-020-2308-7
2. Martorell, L., Nascimento, M., Colome, R. et al. Four sisters compound heterozygotes for the pre- and full mutation in fragile X syndrome and a complete inactivation of X-functional chromosome: implications for genetic counseling. J Hum Genet 56, 87–90 (2011). https://doi.org/10.1038/jhg.2010.140
3. Loureiro, J.R., Oliveira, C.L., Sequeiros, J. et al. A repeat-primed PCR assay for pentanucleotide repeat alleles in spinocerebellar ataxia type 37. J Hum Genet 63, 981–987 (2018). https://doi.org/10.1038/s10038-018-0474-3
4. Egor Dolzhenko, Viraj Deshpande, Felix Schlesinger, Peter Krusche, Roman Petrovski, Sai Chen, Dorothea Emig-Agius, Andrew Gross, Giuseppe Narzisi, Brett Bowman, Konrad Scheffler, Joke J F A van Vugt, Courtney French, Alba Sanchis-Juan, Kristina Ibáñez, Arianna Tucci, Bryan R Lajoie, Jan H Veldink, F Lucy Raymond, Ryan J Taft, David R Bentley, Michael A Eberle, ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions, Bioinformatics, Volume 35, Issue 22, 15 November 2019, Pages 4754–4756, https://doi.org/10.1093/bioinformatics/btz431
5. Dolzhenko, E., Bennett, M.F., Richmond, P.A. et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol 21, 102 (2020). https://doi.org/10.1186/s13059-020-02017-z