简介
近年来,人类基因组研究在表征基因组的暗区方面面临着重大挑战。这些区域由于绘制率低而难以组装或比对,从而导致可定位read很少或没有,并且比对read的定位质量较低。这些暗区通常位于高度多态性或重复的基因组区域,虽然仍难以解析,但却蕴含着宝贵的见解,对于推进我们对人类基因组的理解至关重要1,2。
人类基因组研究中的另一个主要挑战是参考偏差,其源于使用单个单倍体人类参考基因组来表示不同群体中人类序列多样性的局限性3。这两个挑战是相互关联的,因为参考偏差会加剧基因组某些区域read的定位难度。GRCh38参考组装4试图通过引入FASTA参考中原生包含的替代(alt)contig(尤其是在高度多态性区域)来解决至少部分参考偏差问题,以更好地捕获人类遗传变异。这些原生的alt contig代表了主要contig中相应区域的替代路径。然而,处理这些额外的contig需要特别小心,因为处理不当可能会引入比contig试图解决的错误更多的错误。我们在之前的Illumina Genomics Research Hub文章中解释了这一点5,我们展示了DRAGEN能够有效利用这些原生alt contigs与GRCh38参考中主要contig之间的关系来提高定位准确性。最初,它使用alt-aware比对升级程序进一步提高了定位准确性,后来在3.9版本中该程序被alt-masking方法取代。
然而,这些有限的进步尚未完全捕获到大多数的人类变异。为了解决这个问题,研究领域正在创建一组高质量的组装集以供参考。这项工作由多个泛基因组联盟牵头,例如人类泛基因组参考联盟(HPRC)6、中国泛基因组研究联盟(CPC)7和阿拉伯泛基因组参考联盟(APR)8等,旨在创建针对特定祖先的全球性参考集。紧跟HPRC和其他联盟的前进步伐,DRAGEN在3.7版本中首次引入了多基因组定位程序和泛基因组参考的概念,大幅提高了因美纳read在具有挑战性的基因组区域的定位准确性9。
注: 一直以来,我们将执行定位的方法和用作参考的样本集的组合称为“多基因组(图形)参考”。未来,我们将把用于执行定位的方法(多基因组定位程序)与用作定位参考的样本集(泛基因组参考)区分开,以便更好地回顾性描述各版本DRAGEN中引入的更新。
如图1所示,DRAGEN已经陆续发布了几代多基因组定位程序和泛基因组参考,每次迭代都进一步提高了难以比对区域的read定位准确性。图2说明了用于定位的参考的演变。
DRAGEN v3.7中发布的第一代多基因组定位程序使用从16个欧洲样本组成的泛基因组参考中提取的一组群体单倍型扩展了原生alt contig。与使用线性参考(包括原生alt contig)相比,第一代多基因组定位程序的引入使SNP错误减少了47%,indel错误减少了24%。
在DRAGEN v4.2中,我们将泛基因组参考从16个具有欧洲血统的样本扩展到32个具有全球血统的样本。这项改进减少了祖源偏向性,进一步提高了变异检出的准确性。
随着DRAGEN v4.3的发布,我们推出了第二代多基因组定位程序,这使得我们的泛基因组参考从32个扩展到128个群体样本,包含全球26个不同血统(图3)。该泛基因组参考是迄今为止在DRAGEN上推出的最具多样性的群体样本集!
在本文中,我们讨论了使用泛基因组参考的多基因组定位方法的演变,并展示了使用第二代多基因组定位程序和最新的128个样本泛基因组参考时,基因组的所有基准区域和难以比对区域的准确性都有显著提高。
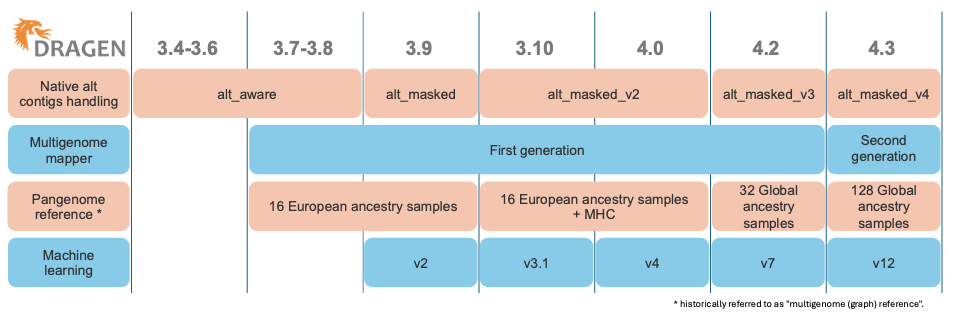
图1:DRAGEN组件的演变,包括原生alt contig处理、多基因组定位程序、泛基因组参考和机器学习。
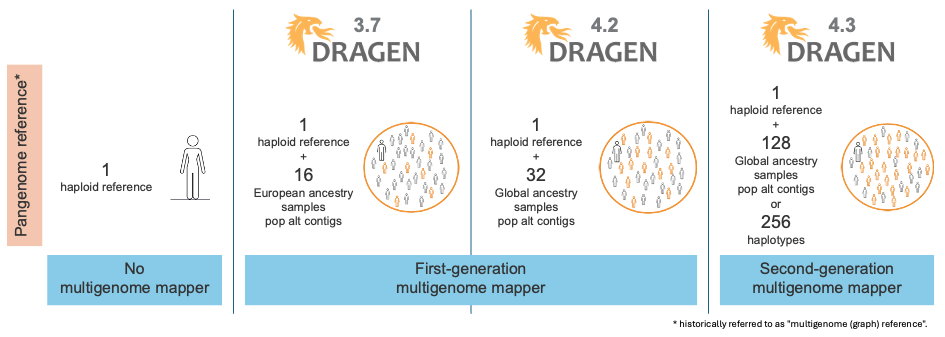
图2:DRAGEN多基因组定位程序的演变——群体感知和泛基因组参考。
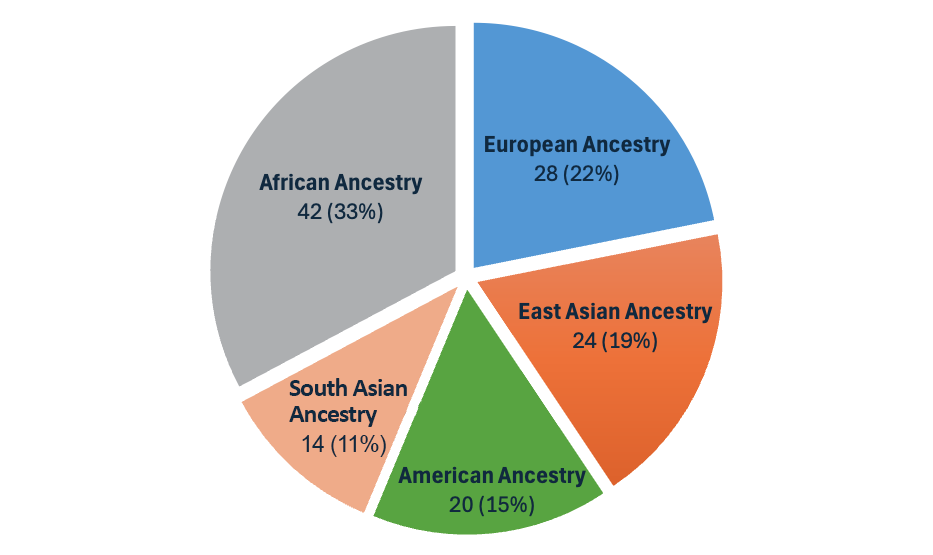
图3:DRAGEN v4.3中的泛基因组参考由来自全球26个血统的128个群体样本组成。
DRAGEN v4.3带来迄今为止最准确的DRAGEN分析
图4显示了连续版本的DRAGEN检测HG002 NIST样本的准确性,覆盖v4.2.1所有基准区域10,组合检测SNP和indel。随着时间的推移,DRAGEN的准确性得到了显著提高,在过去四年中错误率降低了83%。在3.7.5版本中引入了第一代多基因组定位程序和16个样本泛基因组参考,首次大幅降低了错误率。随着DRAGEN v4.3中第二代多基因组定位程序和128个样本泛基因组参考的引入,DRAGEN继续为准确性设定新标准,与DRAGEN v4.2相比,错误率再次大幅降低了40%。
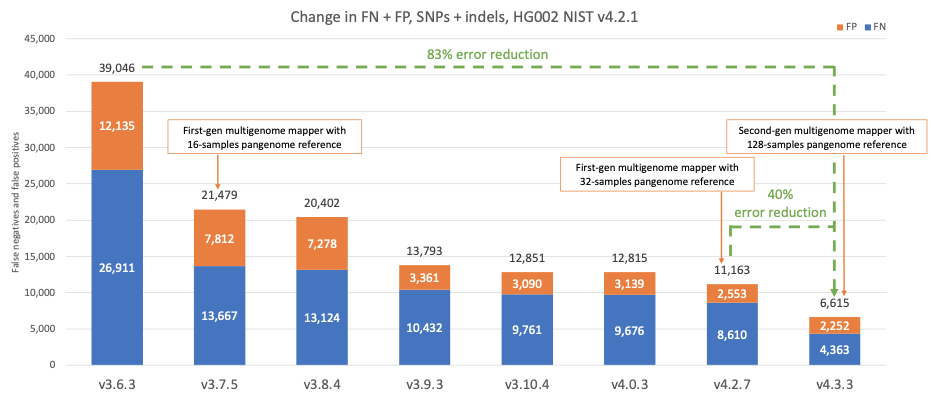
图4:在连续的DRAGEN版本中,HG002 NIST v4.2.1 SNP和indel假阳性和假阴性错误计数。
使用NIST v4.2.1真值集对GIAB样本进行基准测试
在图5中,我们比较了DRAGEN v4.3、DRAGEN v4.2和第三方流程(由用于比对的Giraffe11 1.54.0和用于小变异检出的DeepVariant12 1.6.0组成)的准确性,使用Genome in a Bottle(GIAB)13v4.2.1分析HG001–HG007样本的所有基准区域14。与Giraffe-DeepVariant相比,DRAGEN v4.3在组合SNP和indel上的平均错误率减少了61.61%,其中SNP的平均错误率减少了63.8%,indel的平均错误率减少了53.53%。
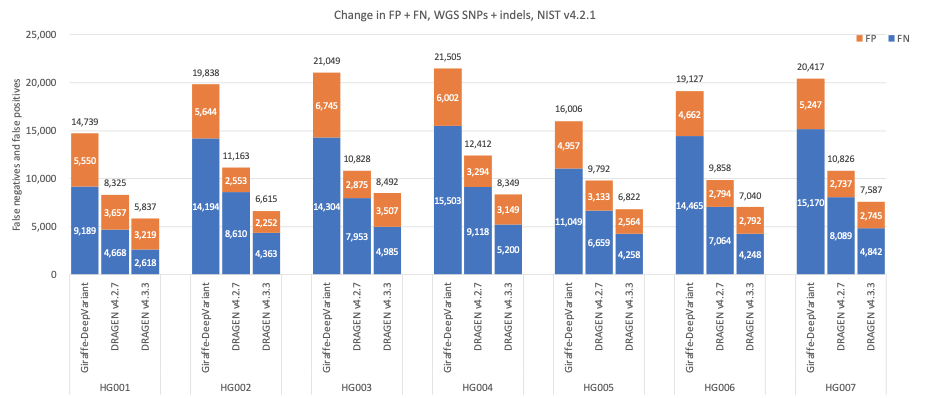
图5:7个GIAB样本(HG001-7)的SNP和indel累积错误计数,使用NIST v4.2.1真值集,DRAGEN v4.2、DRAGEN v4.3和Giraffe-DeepVariant用于分析。
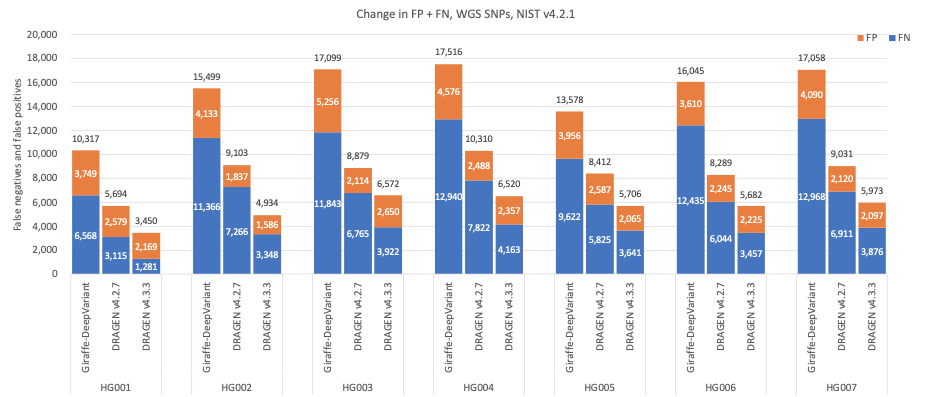
图6:7个GIAB样本(HG001-7)的SNP累积错误计数,使用NIST v4.2.1真值集,DRAGEN v4.2、DRAGEN v4.3和Giraffe-DeepVariant用于分析。
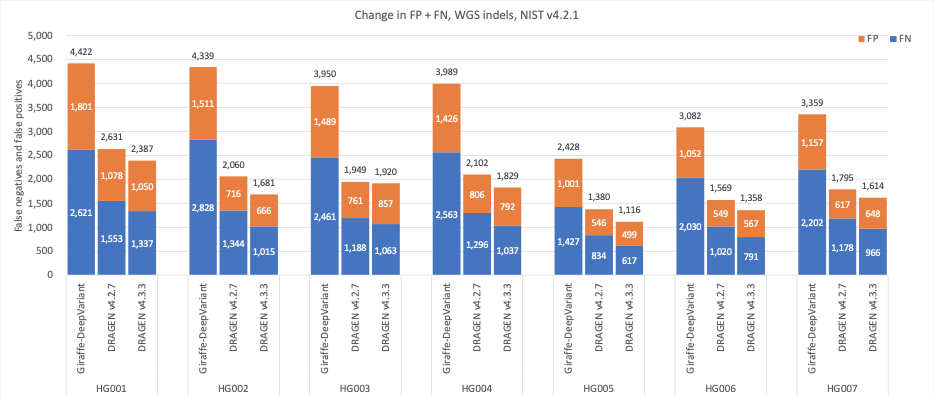
图7:7个GIAB样本(HG001-7)的indel累积错误计数,使用NIST v4.2.1真值集,DRAGEN v4.2、DRAGEN v4.3和Giraffe-DeepVariant用于分析。
对GIAB样本中基因组难以比对区域进行基准测试
图8显示了NIST定义的难以比对区域的准确性结果15,16,DRAGEN 4.3的SNP和indel检测准确性显著提高,与Giraffe-DeepVariant相比,平均错误率减少了65.51%,与DRAGEN 4.2相比减少了37.77%。
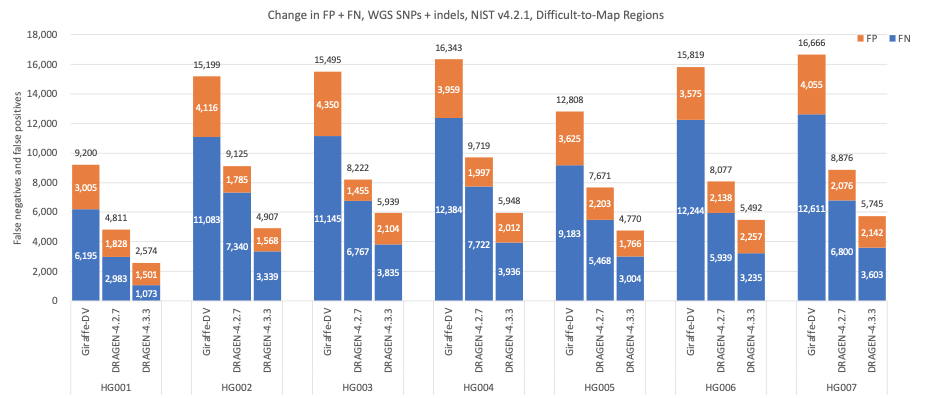
图8:7个GIAB样本(HG001-7)的SNP和indel累积错误计数,使用NIST v4.2.1真值集与难以比对区域BED文件相结合,DRAGEN v4.2、DRAGEN v4.3和Giraffe-DeepVariant用于分析。
使用CMRG真值集对HG002样本进行基准测试
图9显示了连续版本的DRAGEN在复杂医学相关基因(CMRG)真值集中的检测准确性17,组合检测SNP和indel。总错误数随着时间的推移持续减少,这与NIST v4.2.1所有基准区域中的错误数演变一致。最近推出的第二代多基因组定位程序采用了DRAGEN v4.3的128个样本泛基因组参考,使假阳性和假阴性总数额外减少了15%,从而在多个基因中正确检测到85个假阴性检出。DRAGEN v4.2和DRAGEN v4.3的性能均优于Giraffe-DeepVariant流程。与Giraffe-DeepVariant相比,DRAGEN v4.3可正确检测医学相关基因中的200多种额外变异。
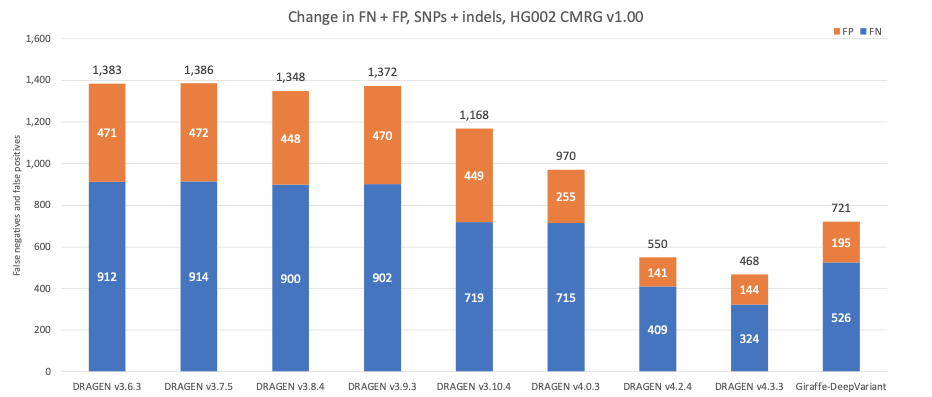
图9:在连续版本的DRAGEN和Giraffe-DeepVariant中使用CMRG真值集所生成的HG002 SNP和indel组合准确性。
DRAGEN第一代多基因组定位方法
对于第一代DRAGEN多基因组定位程序,我们从泛基因组参考样本的定相SNP和indel检出中获得了基因组难以比对区域中的群体单倍型,其中排除了低置信度或低群体等位基因频率变异。特别是,我们从16个欧洲血统样本中得出了32个群体单倍型,从32个全球血统样本中得出了64个群体单倍型。
在参考构建过程中,从这些群体单倍型中衍生出两种类型的泛基因组参考扩展。首先,非定相SNP表示为多核苷酸IUB代码,影响种子定位和比对评分。其次,定相SNP和indel表示为替代序列,每个序列都与主要组装集进行已知liftover比对。read比对以“liftover组”的形式组织,通常包括一个primary-contig成员和一个或多个alt-contig成员。在liftover组级别使用其成员中的最高比对评分进行比对比较、获胜者选择和MAPQ估计。获胜的liftover组的primary-contig成员始终是SAM/BAM输出中报告的成员,因此read定位位置和变异检出仅在标准参考contig中进行。泛基因组参考扩展使定位程序能够更准确地选择标准参考contig比对,并使用群体信息更好地估计MAPQ。变异检出在很大程度上不依赖于泛基因组参考,但受益于提高的定位准确性。因此,该系统从外部看起来像传统的非泛基因组分析,以泛基因组参考作为内部方法来提高准确性。更多详细信息,请参阅我们发表在bioRxiv18上的文章。
DRAGEN第二代多基因组定位方法
DRAGEN v4.3中引入的第二代多基因组定位程序以第一代表现为基础,并解决了其主要局限性。随着可用群体组装数量的增加,第二代多基因组定位程序旨在提高垂直(来自群体泛基因组的样本数量)和水平(全基因组覆盖率)的可扩展性。此外,它依赖于一种允许远程定相的新结构,确保所有群体变异都进行定相。
该数据结构使用专有的压缩方法来避免存储多个单倍型之间共享的冗余变异信息,而只记录局部不同的单倍型集,同时保留所有的远程信息。这种方法能够在添加更多单倍型时实现有效的扩展并提高评分速度。它使单倍型panel的大小显著增加,从DRAGEN v4.2中的64个增加到DRAGEN v4.3中的256个,其中包括更多的血统。
值得注意的是,与包含未定相SNP作为多碱基代码的第一代方法相比,新版本对所有变异进行了定相,从而能够更准确地对单倍型的双端配对进行联合评分。
先前针对替代contig的liftover系统仍在使用,但现在专注于复杂变异。为了提高种子定位过程的灵敏度,群体SNP仍然在主要参考中被编码为多碱基代码,但现在使用来自新数据结构中的定相变异信息来计算比对分数。
命令行选项
之前的Illumina Genomics Research Hub文章中介绍了4.3.3版本之前的DRAGEN运行所使用的命令行选项19。DRAGEN v4.3.3运行使用了以下命令行选项,表1中也对此进行了说明:
dragen \
--fastq-file1 <path-to-R1-fastq> \
--fastq-file2 <path-to-R2-fastq> \
--RGSM HG002 \
–RGID HG002 \
--ref-dir <path-to-reference-directory> \
--output-file-prefix HG002 \
--events-log-file dragen_events.csv \
--output-directory <path-to-output-directory> \
--generate-sa-tags true \
--enable-vcf-compression true \
--enable-variant-caller true \
--enable-map-align true \
--enable-map-align-output true \
--enable-sort true \
--enable-duplicate-marking true \
--enable-bam-indexing true

表1:我们在测试中使用的DRAGEN命令行选项。
DRAGEN v4.3 hg38-alt_masked.graph.cnv.hla.rna_v4哈希表可从DRAGEN产品文件页面获取20,DRAGEN v4.3 ML模型v12.0为默认模型,并与DRAGEN可执行文件组合提供。
使用Nature Biotechnology21中所述的RTG工具包验证与NIST真值的一致性。比较示例命令如下:
java \
-Djava.awt.headless=true \
-Dtalkback=false \
-Dusage=false \
-Xmx40g \
-jar RTG-3.9.1.jar vcfeval \
-b <truth>.vcf.gz \
-c <query>.vcf.gz \
-t <tmp_dir> \
-o <output_dir> \
--output-mode annotate \
--vcf-score-field QUAL \
--bed-regions <truth>.bed \
-Z \
--sample <truth sample>,<query sample> \
--ref-overlap
对于所有DRAGEN运行,我们使用推荐的<sample>.hard-filtered.vcf.gz VCF输出文件,可获得最佳f1评分测量结果。
在评估难以比对区域时,我们还添加了--evaluation-regions<stratification> .bed标志,为RTG工具包提供分层bed。
对于基于Giraffe的流程,我们使用AWS上发布的hprc-v1.1-mc-grch38.d9参考23,遵循DeepVariant-VG案例研究中指定的方案22,并使用Giraffe v1.52.0比对read。命令行和参数如下:
vg giraffe \
--progress \
--read-group "ID:1 LB:lib1 SM:HG002 PL:illumina PU:unit1" \
--sample "HG002" \
--prune-low-cplx \
--max-fragment-length 3000 \
--output-format bam \
-f <path-to-R1-fastq> \
-f <path-to-R2-fastq> \
-x hprc-v1.1-mc-grch38.d9.xg \
-Z hprc-v1.1-mc-grch38.d9.gbz \
-d hprc-v1.1-mc-grch38.d9.dist \
-m hprc-v1.1-mc-grch38.d9.min \
--ref-paths hprc-v1.1-mc-grch38.d9.ref_paths \
-t 32 > HG002.giraffe.grch38.d9.bam
使用sambamba v0.8.1对输出BAM进行排序,并使用samtools v1.15.1进行标签化:
sambamba sort \
-t 32 \
-o HG002.giraffe.grch38.d9.sort.bam \
HG002.giraffe.grch38.d9.bam
samtools index \
-@ 32 \
HG002.giraffe.grch38.d9.sort.bam
使用DeepVariant v1.6.0和以下singularity命令进行变异检出:
singularity run \
--bind "${INPUT_DIR}:/mnt/input,${REF_DIR}:/mnt/reference,${OUTPUT_DIR}:/mnt/output,${BIND_TMPDIR}:/tmp" \
deepvariant_1.6.0.sif \
/opt/deepvariant/bin/run_deepvariant \
--ref="/mnt/reference/hprc-v1.1-mc-grch38.d9.fa" \
--reads="/mnt/input/HG002.giraffe.grch38.d9.sort.bam" \
--model_type="WGS" \
--sample_name="HG002" \
--output_vcf="/mnt/output/HG002.vcf.gz" \
--output_gvcf="/mnt/output/HG002.g.vcf.gz" \
--make_examples_extra_args="min_mapping_quality=1,keep_legacy_allele_counter_behavior=true,normalize_reads=true" \
--haploid_contigs="chrX,chrY" \
--par_regions_bed="/mnt/reference/hprc-v1.1-mc-grch38.d9.par_regions.bed" \
--num_shards="40"
总结
各版本DRAGEN通过精心设计其组件不断为准确性设定新的标准。虽然本文重点介绍了基于泛基因组参考的新一代DRAGEN多基因组定位(第二代),但机器学习和变异检出等其他组件在DRAGEN v4.3中也有所增强,其性能得到进一步提高。
了解有关DRAGEN v4.3改进的更多信息,请访问因美纳软件资源博客。
总体而言,DRAGEN v4.3在基因组暗区的生殖系小变异检测方面所展示的准确性令人印象深刻,在NIST v4.2.1所有基准区域、难以比对区域以及CMRG区域的测试中,其性能始终优于以前的版本和第三方流程。DRAGEN以其准确性、速度、可扩展性、普遍性和全面性而闻名,它仍然是基因组分析的重要工具,以强大的资源支持研究人员和科学家实现全面、高效的基因组数据分析。
参考文献
- Ebbert MTW, Jensen TD, Jansen-West K, et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 2019;20(1):97. doi:10.1186/s13059-019-1707-2
- Ryan NM, Corvin A. Investigating the dark-side of the genome: a barrier to human disease variant discovery? Biol Res. 2023;56(1):42. doi:10.1186/s40659-023-00455-0
- Miga KH, Wang T. The Need for a Human Pangenome Reference Sequence. Annu Rev Genomics Hum Genet. 2021;22:81-102. doi:10.1146/annurev-genom-120120-081921
- GRCh38 Full Analysis Set Plus Decoys HLA. ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa
- Catreux S, Farrell F, Mehio R, et al. Demystifying the versions of GRCh38/hg38 reference genomes, how they are used in DRAGEN and their impact on accuracy. Illumina website. illumina.com/science/genomics-research/articles/dragen-demystifying-reference-genomes.html. Published December 9, 2021. Accessed July 18, 2024.
- Liao WW, Asri M, Ebler J, et al. A draft human pangenome reference. Nature. 2023;617(7960)312-324. doi:10.1038/s41586-023-05896-x
- Gao Y, Yang X, Chen H, et al. A pangenome reference of 36 Chinese populations. Nature. 2023;619(7968):112-121. doi:10.1038/s41586-023-06173-7
- Uddin M, Nassir N, Almarri M, et al. A draft Arab pangenome reference. Preprint. Research Square. October 2023. doi:10.21203/rs.3.rs-3490341/v1
- Catreux S, Jain V, Murray L, et al. DRAGEN sets new standard for data accuracy in PrecisionFDA benchmark data. Optimizing variant calling performance with Illumina machine learning and DRAGEN graph. Illumina website. illumina.com/science/genomics-research/articles/dragen-shines-again-precisionfda-truth-challenge-v2.html. Published January 12, 2022. Accessed July 18, 2024.
- GIAB HG002 v4.2.1 truth. ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/AshkenazimTrio/HG002_NA24385_son/NISTv4.2.1/.
- Sirén J, Monlong J, Chang X, et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science. 2021;374(6574). doi:10.1126/science.abg8871
- Poplin R, Chang PC, Alexander D, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36(10):983-987. doi:10.1038/nbt.4235
- Wagner J, Olson ND, Harris L, et al. Benchmarking challenging small variants with linked and long reads. Cell Genom. 2022;2(5):100128. doi:10.1016/j.xgen.2022.100128
- GIAB samples release. ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/.
- Difficult-to-Map Regions BED file. ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/genome-stratifications/v3.3/GRCh38@all/Union/GRCh38_alllowmapandsegdupregions.bed.gz.
- Olson ND, Wagner J, McDaniel J, et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genom. 2022;2(5):100129. doi:10.1016/j.xgen.2022.100129
- Wagner J, Olson ND, Harris L, et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat Biotechnol. 2022;40(5):672-680. doi:10.1038/s41587-021-01158-1
- Behera S, Catreux S, Rossi M, et al. Comprehensive and accurate genome analysis at scale using DRAGEN accelerated algorithms. Preprint. bioRxiv. 2024;2024.01.02.573821. doi:10.1101/2024.01.02.573821
- Rossi M, Catreux S, Roddey C, et al. Unlocking the full potential of Illumina genomes: The journey to enhanced variant calling quality with DRAGEN informatics and high-quality sequencing. Illumina website. illumina.com/science/genomics-research/articles/CMRG_hg38.html. Published June 29, 2023. Accessed July 18, 2024.
- DRAGEN Bio-IT Platform Product Files. emea.support.illumina.com/sequencing/sequencing_software/dragen-bio-it-platform/product_files.html.
- Krusche P, Trigg L, Boutros PC, et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat Biotechnol. 2019;37(5):555-560. doi:10.1038/s41587-019-0054-x
- Using graph genomes: VG Giraffe + DeepVariant case study. github.com/google/deepvariant/blob/0bba6a71b8b0c2046a3b01c0bda1a5a0d2b80fca/docs/deepvariant-vg-case-study.md.
- hprc-v1.1-mc-grch38. s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=pangenomes/freeze/freeze1/minigraph-cactus/hprc-v1.1-mc-grch38/.